Li Xiaoyang, Zheng Yongsheng, Li Shaoyuan, Nair Umesh, Sun Chong, Zhao Chongbo, Lu Jiahong, Zhang Victor Wei, Maljevic Snezana, Petrou Steven, Lin Jie
Department of Neurology, Huashan Hospital, Fudan University, Shanghai, China.
Department of Neurology, University of North Carolina, Chapel Hill, USA.
Ann Transl Med. 2021 Sep;9(18):1397. doi: 10.21037/atm-21-1885.
KCNC1 encodes Kv3.1, a subunit of the Kv3 voltage-gated potassium channels. It is predominantly expressed in inhibitory GABAergic interneurons and cerebellar neurons. Kv3.1 channelopathy has been linked to a variety of human diseases including epilepsy, developmental delay, and ataxia. Characterization of structural and functional disturbances of this channel, and its relationship to a heterogenous group of clinical phenotypes, is a current topic of research. We herein characterize the clinical phenotype as well as the functional and structural consequences of the novel p.R317S variant. We further set out to explore the mechanistic basis for the spectrum of related channelopathies.
Variant was identified via whole-exome sequencing and its functional impact was determined using two-electrode voltage clamp recordings in oocytes. Homolog modeling and structural analysis were performed on the p.R317S variant and other related variants.
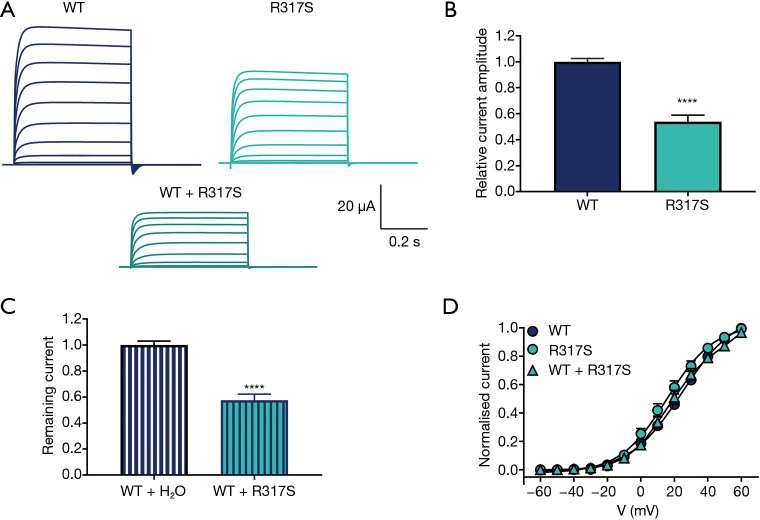
We identified a novel loss-of-function KCNC1 variant c.949C>A (p.R317S) presenting with symptoms similar to myoclonic epilepsy and ataxia due to potassium channel (MEAK), but with distinct radiological features. Functional analysis in the oocyte's expression system revealed that the current amplitudes were significantly decreased in the p.R317S variant compared to the wild type, indicating a dominant-negative effect. Atomic structural analysis of the related variants provided a possible mechanistic explanation for the heterogeneity in the clinical spectrum.
We have identified the p.R317S loss-of-function variant in the gene, expanded the spectrum of potassium channelopathy and provided mechanistic insights into related disorders.
KCNC1基因编码Kv3.1,它是Kv3电压门控钾通道的一个亚基。它主要在抑制性γ-氨基丁酸能中间神经元和小脑神经元中表达。Kv3.1通道病与多种人类疾病有关,包括癫痫、发育迟缓及共济失调。对该通道结构和功能紊乱的特征及其与一组异质性临床表型的关系进行研究是当前的一个课题。我们在此描述了新型p.R317S变异的临床表型以及功能和结构后果。我们进一步着手探索相关通道病谱系的机制基础。
通过全外显子测序鉴定变异,并使用卵母细胞双电极电压钳记录来确定其功能影响。对p.R317S变异及其他相关变异进行同源建模和结构分析。
我们鉴定出一种新型功能缺失的KCNC1变异c.949C>A(p.R317S),其表现出与钾通道相关的肌阵挛性癫痫和共济失调(MEAK)相似的症状,但具有独特的影像学特征。在卵母细胞表达系统中的功能分析显示,与野生型相比,p.R317S变异的电流幅度显著降低,表明存在显性负效应。对相关变异的原子结构分析为临床谱系的异质性提供了一种可能的机理解释。
我们在该基因中鉴定出p.R317S功能缺失变异,扩展了钾通道病的谱系,并为相关疾病提供了机制性见解。