Toulouse Biotechnology Institute, Université de Toulouse, CNRS, INRAE, INSA, ANITI, 31077 Toulouse, France.
Université Fédérale de Toulouse, ANITI, INRAE, UR 875, 31326 Toulouse, France.
Int J Mol Sci. 2021 Oct 29;22(21):11741. doi: 10.3390/ijms222111741.
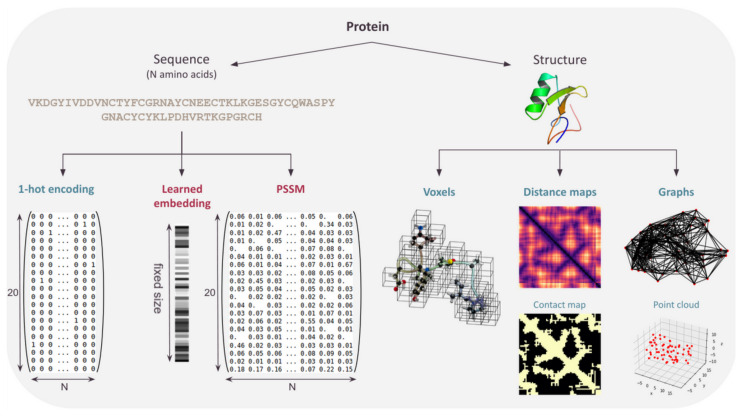
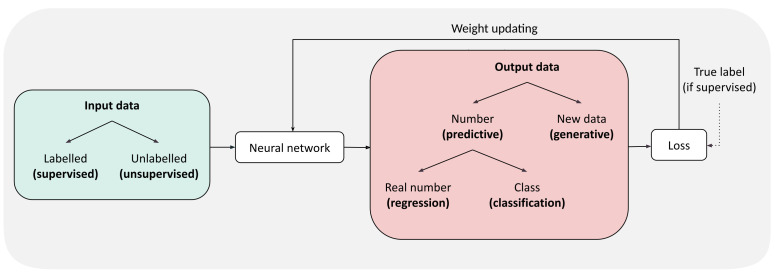
Computational Protein Design (CPD) has produced impressive results for engineering new proteins, resulting in a wide variety of applications. In the past few years, various efforts have aimed at replacing or improving existing design methods using Deep Learning technology to leverage the amount of publicly available protein data. Deep Learning (DL) is a very powerful tool to extract patterns from raw data, provided that data are formatted as mathematical objects and the architecture processing them is well suited to the targeted problem. In the case of protein data, specific representations are needed for both the amino acid sequence and the protein structure in order to capture respectively 1D and 3D information. As no consensus has been reached about the most suitable representations, this review describes the representations used so far, discusses their strengths and weaknesses, and details their associated DL architecture for design and related tasks.
计算蛋白质设计(CPD)在工程新蛋白质方面取得了令人印象深刻的成果,产生了各种各样的应用。在过去的几年中,各种努力旨在使用深度学习技术取代或改进现有的设计方法,以利用大量可用的蛋白质数据。深度学习(DL)是从原始数据中提取模式的非常强大的工具,前提是数据被格式化为数学对象,并且处理它们的架构适合于目标问题。在蛋白质数据的情况下,需要对氨基酸序列和蛋白质结构进行特定的表示,以便分别捕获 1D 和 3D 信息。由于关于最合适的表示形式尚未达成共识,因此本综述描述了迄今为止使用的表示形式,讨论了它们的优缺点,并详细介绍了它们用于设计和相关任务的相关深度学习架构。