Liu Xuhan, Ye Kai, van Vlijmen Herman W T, Emmerich Michael T M, IJzerman Adriaan P, van Westen Gerard J P
Drug Discovery and Safety, Leiden Academic Centre for Drug Research, Einsteinweg 55, 2333 CC, Leiden, The Netherlands.
School of Electronics and Information Engineering, Xi'an Jiaotong University, 28 Xianning W Rd, Xi'an, China.
J Cheminform. 2021 Nov 12;13(1):85. doi: 10.1186/s13321-021-00561-9.
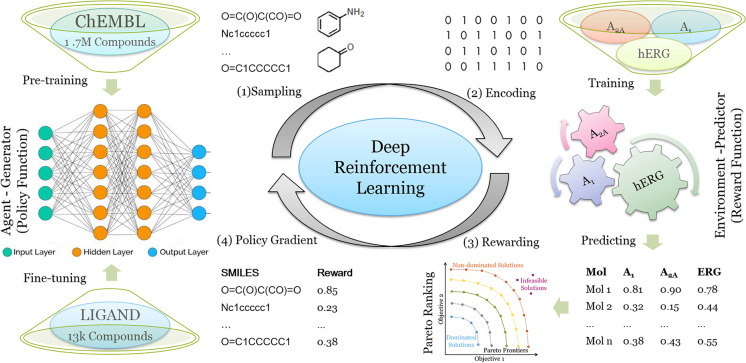
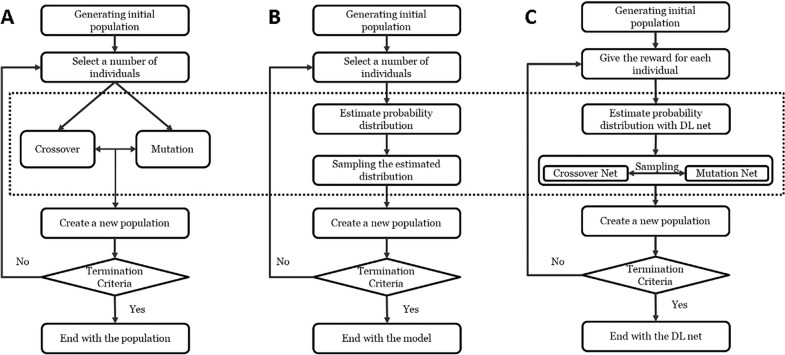
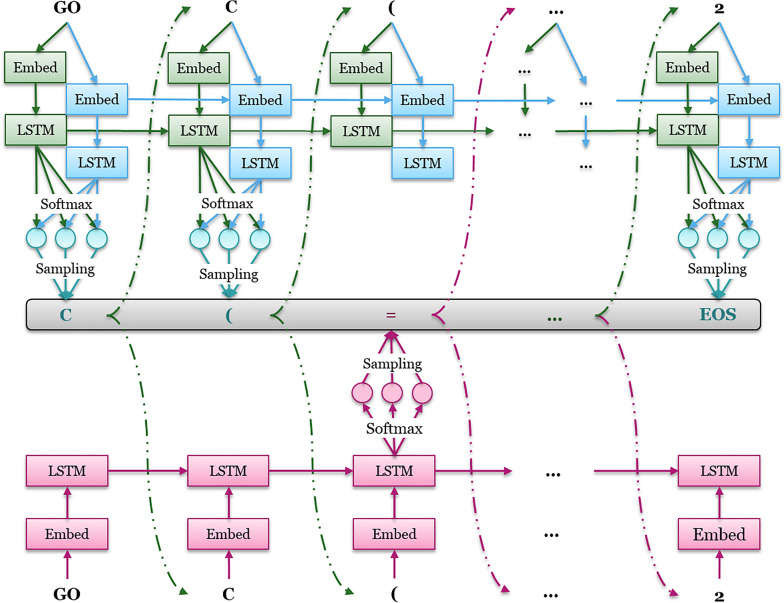
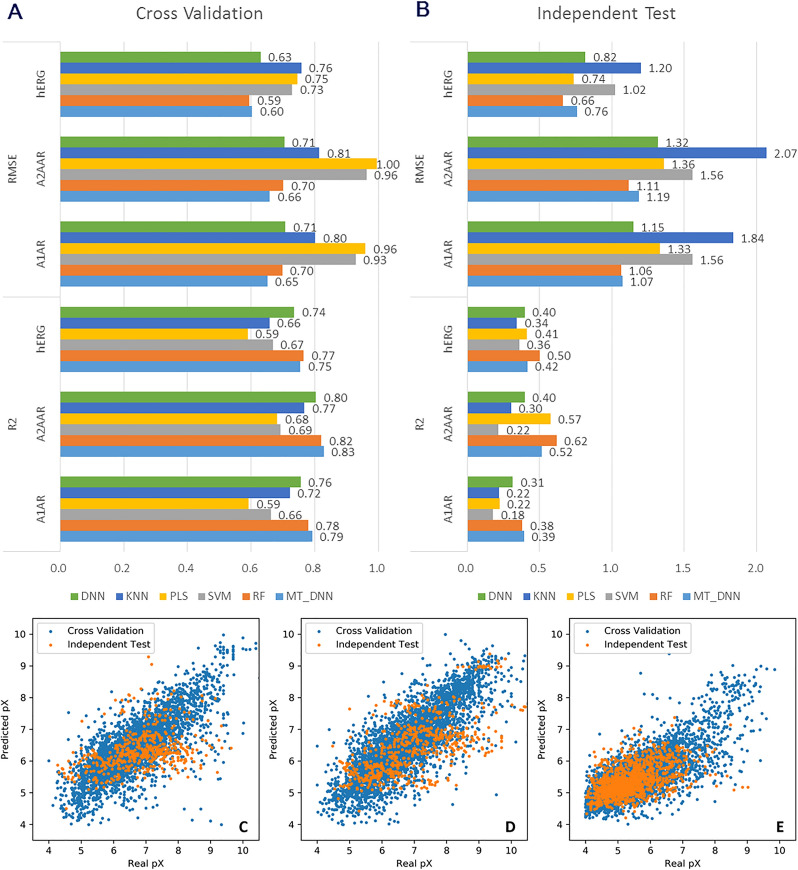
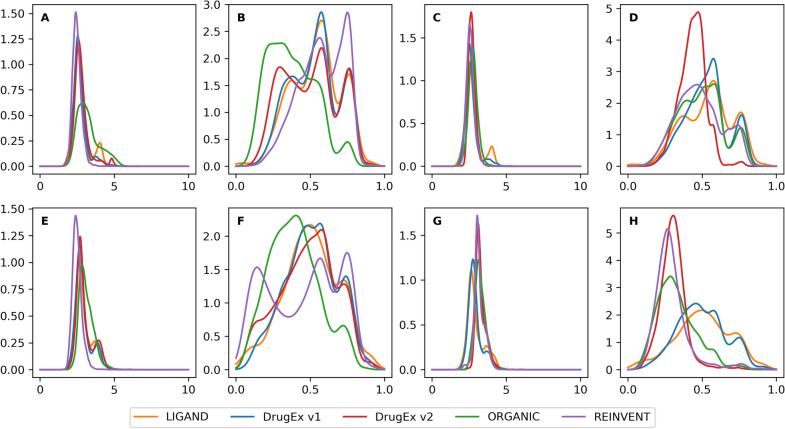
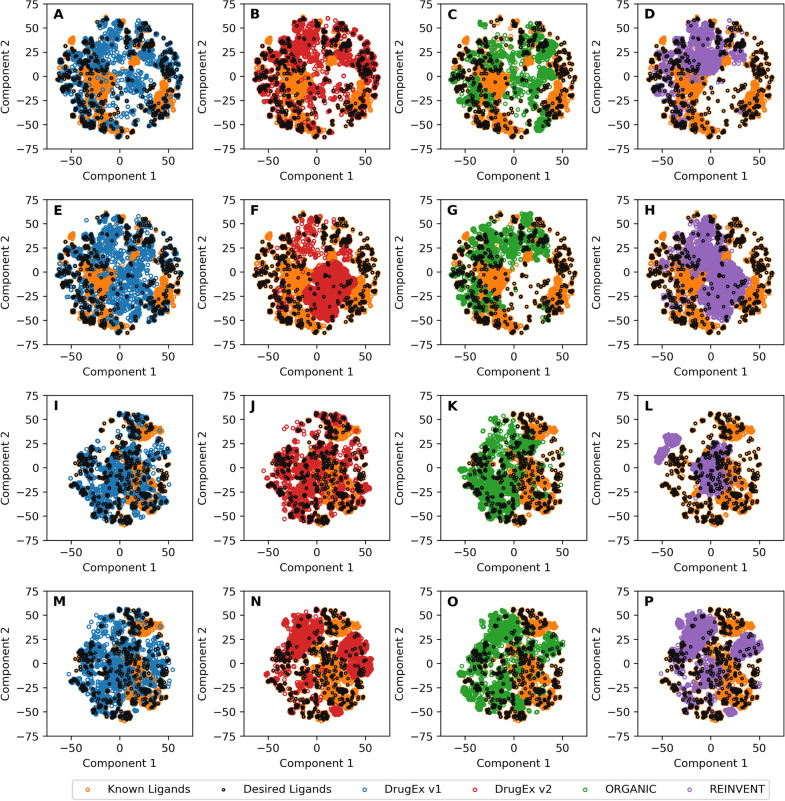
In polypharmacology drugs are required to bind to multiple specific targets, for example to enhance efficacy or to reduce resistance formation. Although deep learning has achieved a breakthrough in de novo design in drug discovery, most of its applications only focus on a single drug target to generate drug-like active molecules. However, in reality drug molecules often interact with more than one target which can have desired (polypharmacology) or undesired (toxicity) effects. In a previous study we proposed a new method named DrugEx that integrates an exploration strategy into RNN-based reinforcement learning to improve the diversity of the generated molecules. Here, we extended our DrugEx algorithm with multi-objective optimization to generate drug-like molecules towards multiple targets or one specific target while avoiding off-targets (the two adenosine receptors, AAR and AAR, and the potassium ion channel hERG in this study). In our model, we applied an RNN as the agent and machine learning predictors as the environment. Both the agent and the environment were pre-trained in advance and then interplayed under a reinforcement learning framework. The concept of evolutionary algorithms was merged into our method such that crossover and mutation operations were implemented by the same deep learning model as the agent. During the training loop, the agent generates a batch of SMILES-based molecules. Subsequently scores for all objectives provided by the environment are used to construct Pareto ranks of the generated molecules. For this ranking a non-dominated sorting algorithm and a Tanimoto-based crowding distance algorithm using chemical fingerprints are applied. Here, we adopted GPU acceleration to speed up the process of Pareto optimization. The final reward of each molecule is calculated based on the Pareto ranking with the ranking selection algorithm. The agent is trained under the guidance of the reward to make sure it can generate desired molecules after convergence of the training process. All in all we demonstrate generation of compounds with a diverse predicted selectivity profile towards multiple targets, offering the potential of high efficacy and low toxicity.
在多靶点药理学中,药物需要与多个特定靶点结合,例如以增强疗效或减少耐药性的形成。尽管深度学习在药物发现的从头设计方面取得了突破,但其大多数应用仅专注于单个药物靶点以生成类药物活性分子。然而,在现实中,药物分子通常会与多个靶点相互作用,这可能会产生期望的(多靶点药理学)或不期望的(毒性)效应。在之前的一项研究中,我们提出了一种名为DrugEx的新方法,该方法将探索策略集成到基于循环神经网络(RNN)的强化学习中,以提高生成分子的多样性。在此,我们通过多目标优化扩展了我们的DrugEx算法,以生成针对多个靶点或一个特定靶点的类药物分子,同时避免脱靶效应(本研究中的两个腺苷受体,AAR和AAR,以及钾离子通道hERG)。在我们的模型中,我们将RNN用作智能体,将机器学习预测器用作环境。智能体和环境均预先进行训练,然后在强化学习框架下相互作用。进化算法的概念被融入到我们的方法中,使得交叉和变异操作由与智能体相同的深度学习模型实现。在训练循环中,智能体生成一批基于SMILES的分子。随后,环境提供的所有目标的分数用于构建所生成分子的帕累托排名。为此排名,应用了一种非支配排序算法和一种使用化学指纹的基于塔尼莫托系数的拥挤距离算法。在此,我们采用GPU加速来加快帕累托优化的过程。每个分子的最终奖励基于使用排名选择算法的帕累托排名来计算。智能体在奖励的指导下进行训练,以确保在训练过程收敛后能够生成期望的分子。总而言之,我们展示了针对多个靶点生成具有多样预测选择性谱的化合物,具有高疗效和低毒性的潜力。