Department for Computational Biology of Infection Research, Helmholtz Centre for Infection Research, 38124, Braunschweig, Germany.
Braunschweig Integrated Centre of Systems Biology (BRICS), Technische Universität Braunschweig, 38106, Braunschweig, Germany.
Nat Commun. 2021 Nov 18;12(1):6744. doi: 10.1038/s41467-021-26938-w.
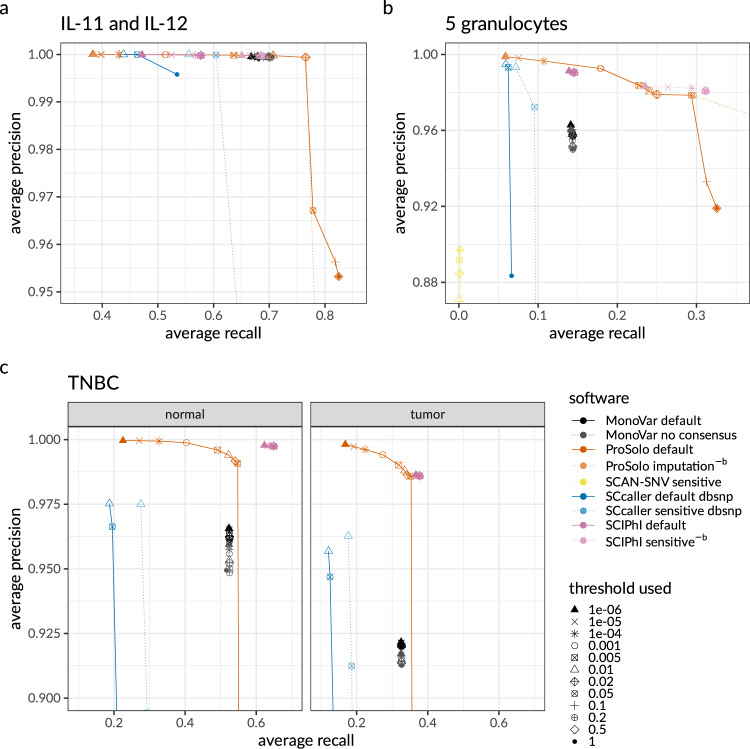
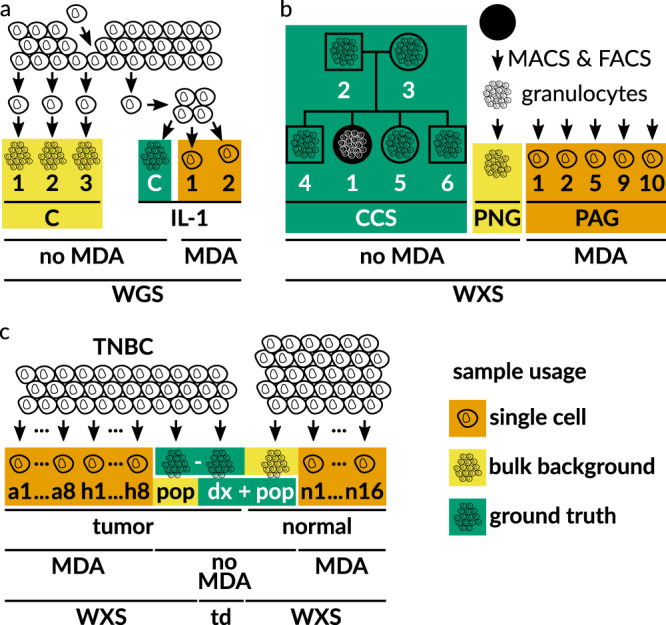
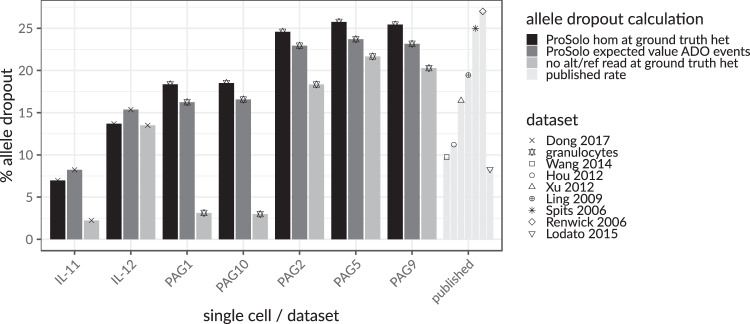
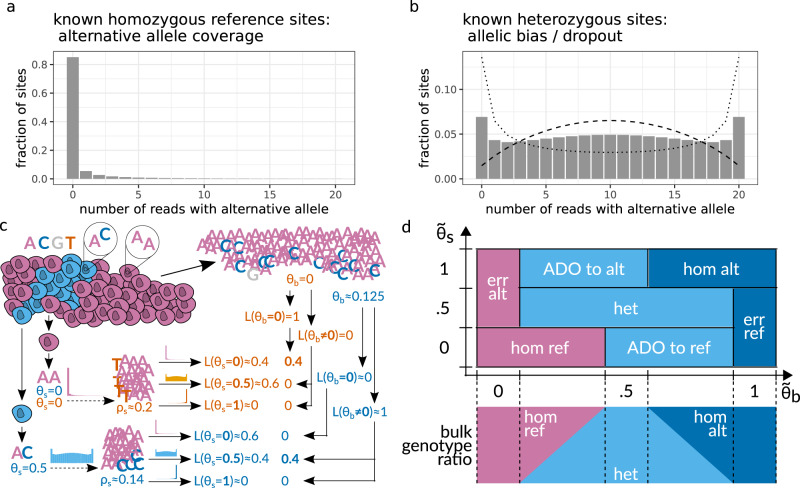
Accurate single cell mutational profiles can reveal genomic cell-to-cell heterogeneity. However, sequencing libraries suitable for genotyping require whole genome amplification, which introduces allelic bias and copy errors. The resulting data violates assumptions of variant callers developed for bulk sequencing. Thus, only dedicated models accounting for amplification bias and errors can provide accurate calls. We present ProSolo for calling single nucleotide variants from multiple displacement amplified (MDA) single cell DNA sequencing data. ProSolo probabilistically models a single cell jointly with a bulk sequencing sample and integrates all relevant MDA biases in a site-specific and scalable-because computationally efficient-manner. This achieves a higher accuracy in calling and genotyping single nucleotide variants in single cells in comparison to state-of-the-art tools and supports imputation of insufficiently covered genotypes, when downstream tools cannot handle missing data. Moreover, ProSolo implements the first approach to control the false discovery rate reliably and flexibly. ProSolo is implemented in an extendable framework, with code and usage at: https://github.com/prosolo/prosolo.
准确的单细胞突变谱可以揭示基因组的细胞间异质性。然而,适合基因分型的测序文库需要全基因组扩增,这会引入等位基因偏倚和拷贝错误。由此产生的数据违反了为批量测序开发的变异调用器的假设。因此,只有专门考虑扩增偏差和错误的模型才能提供准确的调用。我们提出了 ProSolo,用于从多重置换扩增 (MDA) 单细胞 DNA 测序数据中调用单核苷酸变体。ProSolo 联合批量测序样本对单个细胞进行概率建模,并以特定于位点且可扩展的方式(因为计算效率高)整合所有相关的 MDA 偏差。与最先进的工具相比,这在调用和基因分型单细胞中的单核苷酸变体方面实现了更高的准确性,并支持下游工具无法处理缺失数据时对覆盖不足的基因型进行推断。此外,ProSolo 实现了第一个可靠且灵活地控制假发现率的方法。ProSolo 是在可扩展的框架中实现的,代码和用法在:https://github.com/prosolo/prosolo。