Department of Biomedical Informatics, Harvard Medical School, Boston, MA, USA.
Ludwig Center at Harvard, Boston, MA, USA.
Nat Commun. 2019 Aug 29;10(1):3908. doi: 10.1038/s41467-019-11857-8.
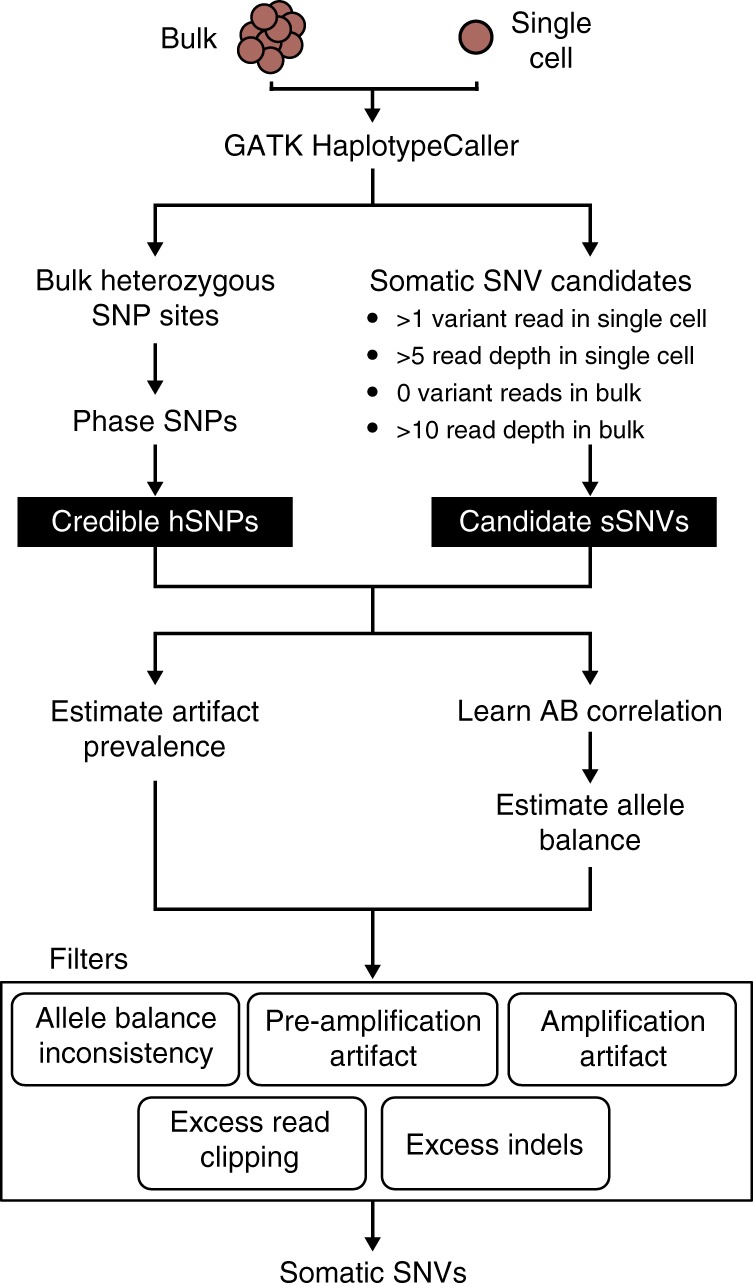
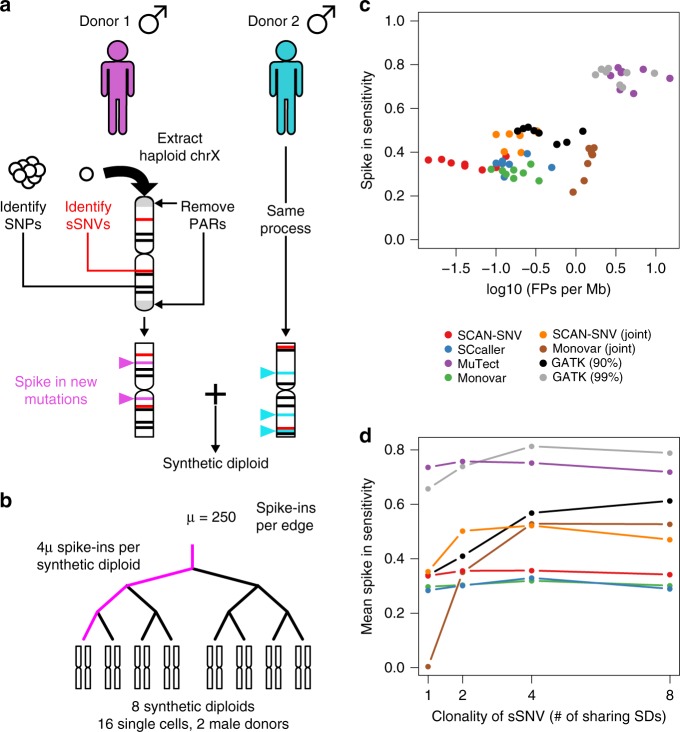
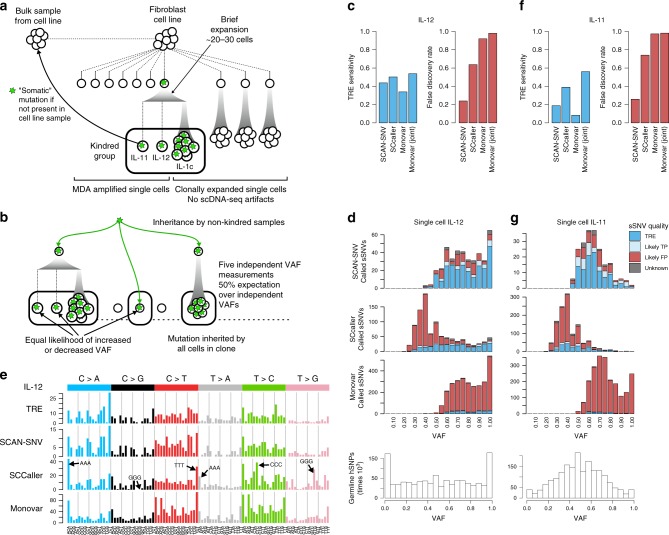
Recent advances in single cell technology have enabled dissection of cellular heterogeneity in great detail. However, analysis of single cell DNA sequencing data remains challenging due to bias and artifacts that arise during DNA extraction and whole-genome amplification, including allelic imbalance and dropout. Here, we present a framework for statistical estimation of allele-specific amplification imbalance at any given position in single cell whole-genome sequencing data by utilizing the allele frequencies of heterozygous single nucleotide polymorphisms in the neighborhood. The resulting allelic imbalance profile is critical for determining whether the variant allele fraction of an observed mutation is consistent with the expected fraction for a true variant. This method, implemented in SCAN-SNV (Single Cell ANalysis of SNVs), substantially improves the identification of somatic variants in single cells. Our allele balance framework is broadly applicable to genotype analysis of any variant type in any data that might exhibit allelic imbalance.
单细胞技术的最新进展使我们能够非常详细地解析细胞异质性。然而,由于在 DNA 提取和全基因组扩增过程中出现的偏倚和伪影,包括等位基因失衡和缺失,单细胞 DNA 测序数据的分析仍然具有挑战性。在这里,我们提出了一种通过利用附近杂合单核苷酸多态性的等位基因频率来统计估计单细胞全基因组测序数据中任何给定位置的等位基因特异性扩增失衡的框架。由此产生的等位基因失衡谱对于确定观察到的突变的变异等位基因分数是否与真实变体的预期分数一致至关重要。这种方法在 SCAN-SNV(单核苷酸变异的单细胞分析)中得到了实现,可大大提高单细胞中体细胞变异的识别能力。我们的等位基因平衡框架广泛适用于任何可能表现出等位基因失衡的数据中任何变体类型的基因型分析。