Wu Shi, Pang Rui, Huang Jiahui, Zhang Feng, Cai Zhihe, Zhang Jumei, Chen Moutong, Xue Liang, Gu Qihui, Wang Juan, Ding Yu, Wan Qiang, Wu Qingping
Guangdong Provincial Key Laboratory of Microbial Safety and Health, State Key Laboratory of Applied Microbiology Southern China, Institute of Microbiology, Guangdong Academy of Sciences, Guangzhou, China.
Guangdong Huankai Microbial Science and Technology Co. Ltd., Guangzhou, China.
Front Microbiol. 2021 Nov 19;12:769642. doi: 10.3389/fmicb.2021.769642. eCollection 2021.
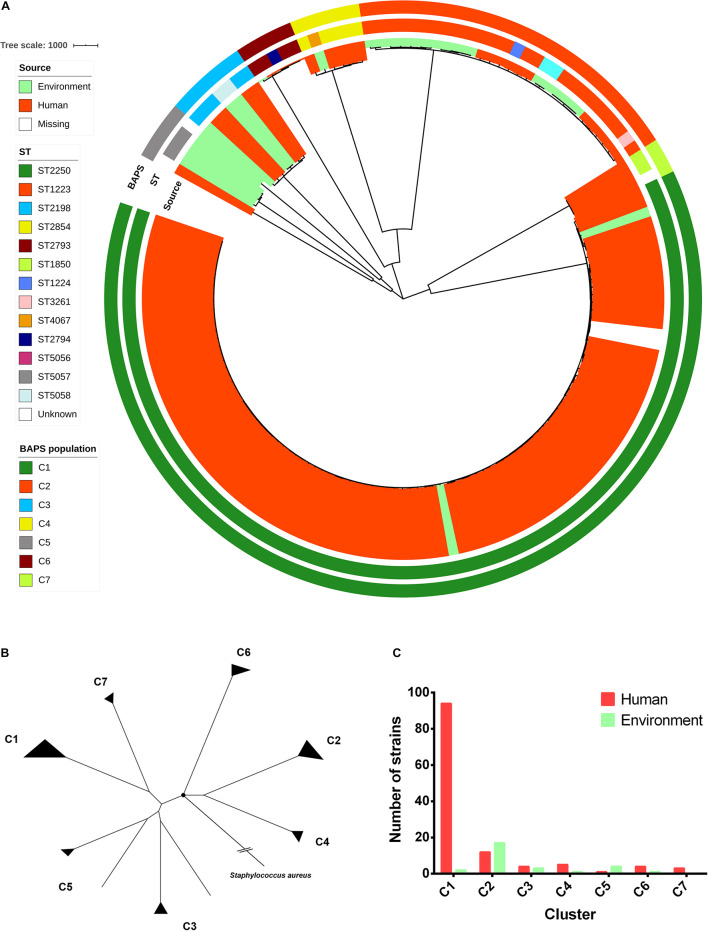
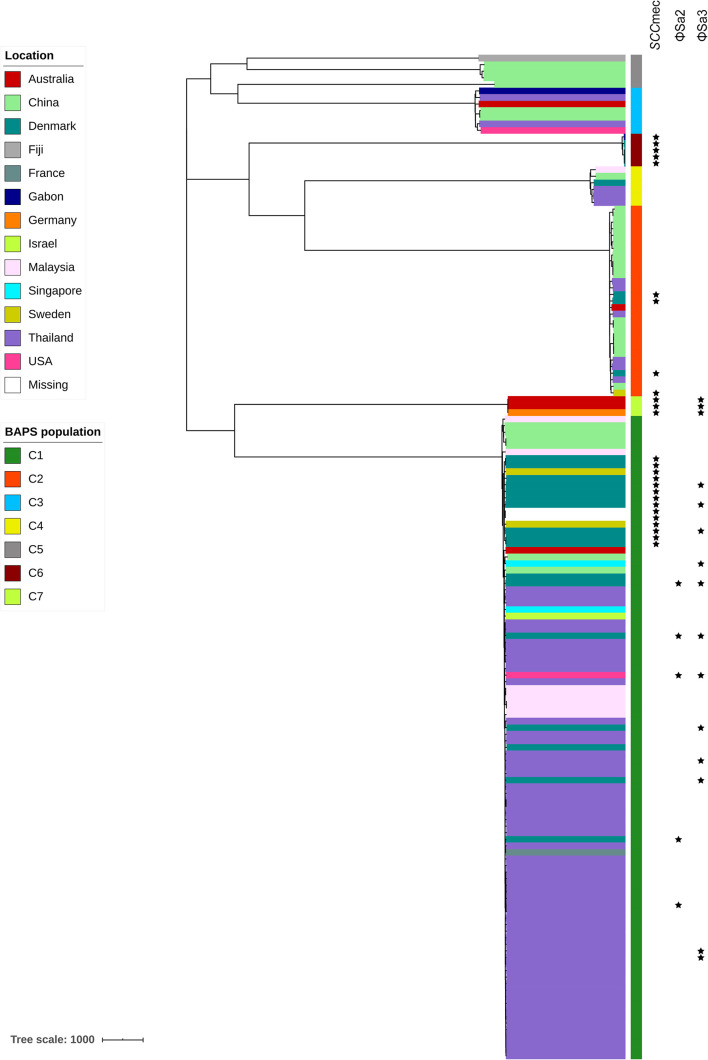
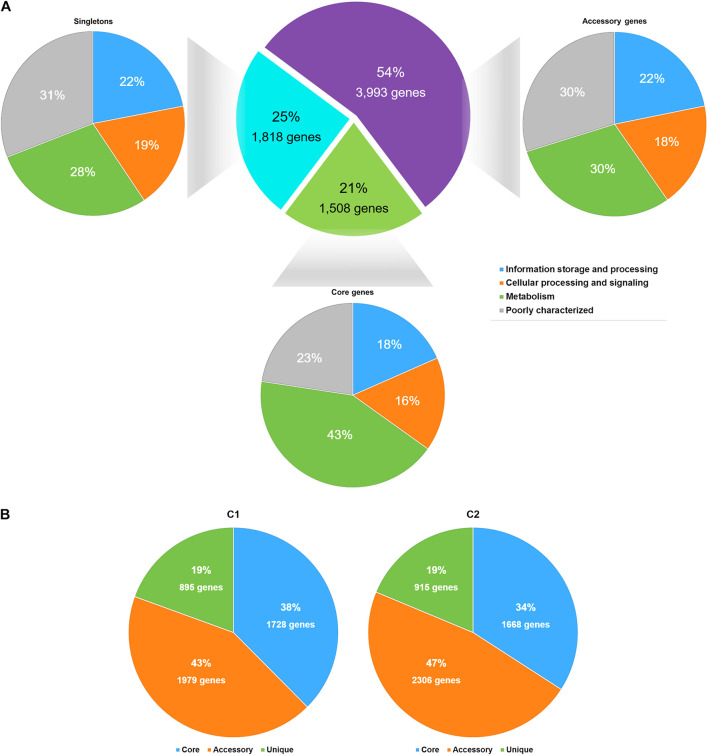
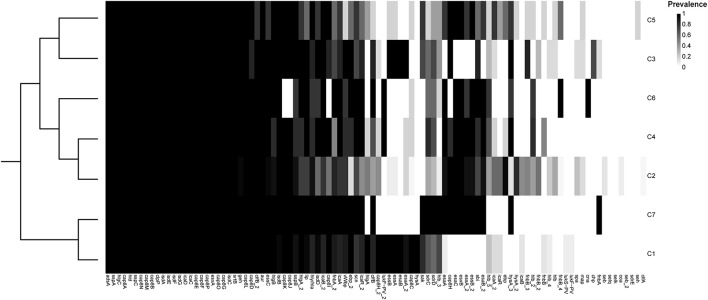
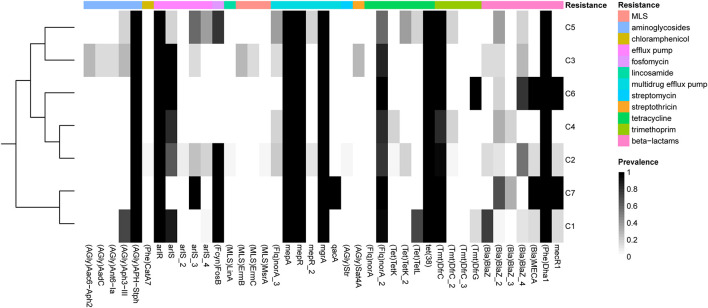
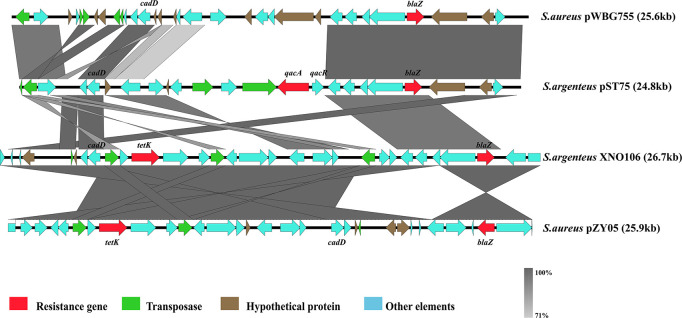
Currently, invasive infections caused by , which is a recently named staphylococcal species, are increasingly reported worldwide. However, only a few genomic studies of . have offered comprehensive information regarding its genetic diversity, epidemiological characteristics, antimicrobial resistance genes (ARGs), virulence genes and other profiles. Here, we describe a comparative genomic analysis by population structure, pangenome, panmobilome, region-specific accessory genes confer an adaptive advantage in 153 . strains which comprised 24 strains sequenced in this study and 129 strains whose genome sequences were available from GenBank. As a result, the population of . comprised seven genetically distinct clades, including two major clades (C1 and C2), with distinct isolation source patterns. Pangenome analysis revealed that . has an open pangenome composed of 7,319 genes and a core genome composed of 1,508 genes. We further determined the distributions of 75 virulence factors (VFs) and 30 known ARGs and identified at least four types of plasmids and 93 complete or partial putative prophages. It indicate that . may show a similar level of pathogenicity to that of . . This study also provides insights into the evolutionary divergence of this pathogen, indicating that the geographical distribution was a potential driving force behind the evolutionary divergence of . . The preferential horizontal acquisition of particular elements, such as staphylococcal cassette chromosome elements and plasmids, was observed in specific regions, revealing potential gene exchange between . strains and local . strains. Moreover, multiple specific genes related to environmental adaptation were identified in strains isolated from East Asia. However, these findings may help promote our understanding of the evolutionary divergence of this bacterium at a high genetic resolution by providing insights into the epidemiology of . and may help combat its spread.
目前,由一种最近命名的葡萄球菌引起的侵袭性感染在全球范围内的报道日益增多。然而,关于该葡萄球菌的基因组研究较少,仅有少数研究提供了关于其遗传多样性、流行病学特征、抗菌耐药基因(ARGs)、毒力基因及其他特征的全面信息。在此,我们对153株该葡萄球菌进行了群体结构、泛基因组、泛移动基因组以及区域特异性辅助基因的比较基因组分析,其中包括本研究测序的24株菌株以及从GenBank获取基因组序列的129株菌株。结果显示,该葡萄球菌群体包含七个遗传上不同的分支,包括两个主要分支(C1和C2),具有不同的分离源模式。泛基因组分析表明,该葡萄球菌具有一个由7319个基因组成的开放泛基因组和一个由1508个基因组成的核心基因组。我们进一步确定了75种毒力因子(VFs)和30种已知ARGs的分布,并鉴定出至少四种类型的质粒和93个完整或部分推定的原噬菌体。这表明该葡萄球菌可能表现出与另一种葡萄球菌相似的致病水平。本研究还深入了解了这种病原体的进化分歧,表明地理分布是该葡萄球菌进化分歧背后的潜在驱动力。在特定区域观察到特定元件(如葡萄球菌盒式染色体元件和质粒)的优先水平获取,揭示了该葡萄球菌菌株与当地其他葡萄球菌菌株之间潜在的基因交换。此外,在从东亚分离的菌株中鉴定出多个与环境适应相关的特定基因。然而,这些发现可能有助于通过深入了解该葡萄球菌的流行病学,以高遗传分辨率促进我们对这种细菌进化分歧的理解,并可能有助于对抗其传播。