Basheer Atia, Zahoor Imran
Genetics and Genomic Laboratory, Department of Animal Breeding and Genetics, University of Veterinary and Animal Sciences, Ravi Campus, Pattoki 55300, Pakistan.
Microorganisms. 2021 Dec 17;9(12):2609. doi: 10.3390/microorganisms9122609.
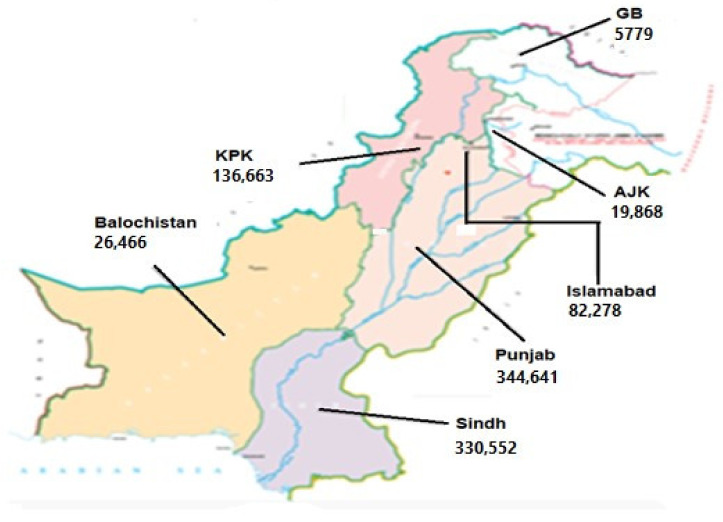
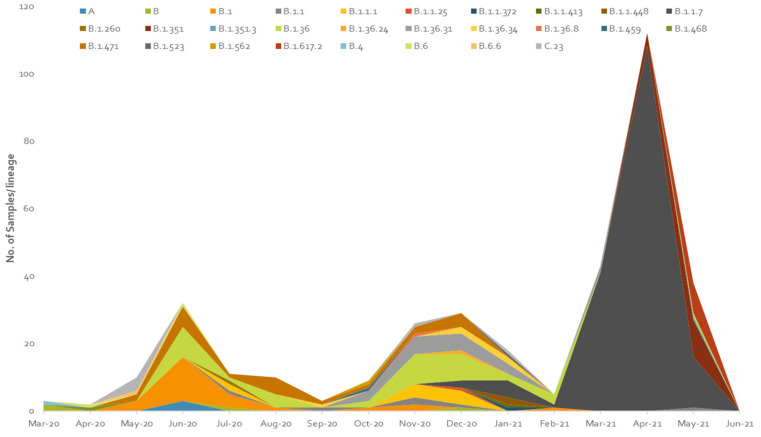
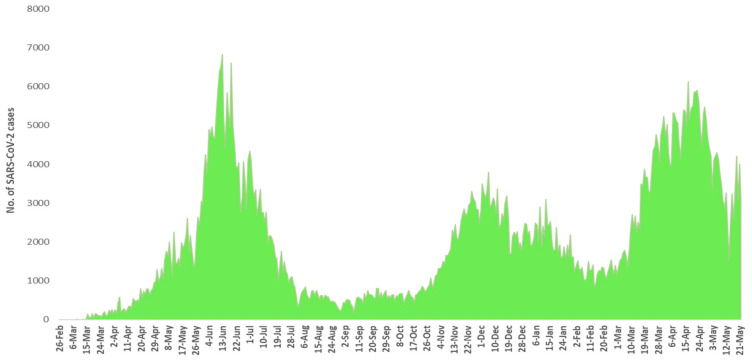
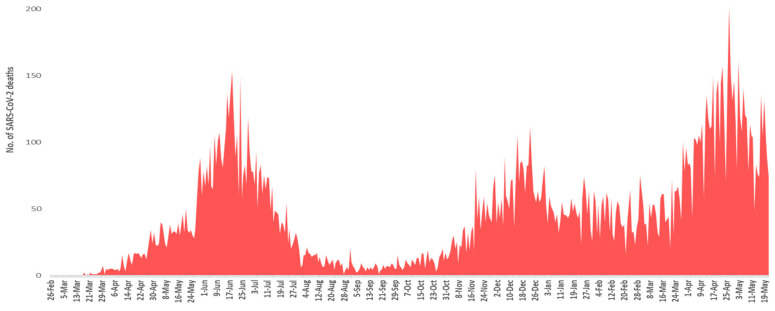
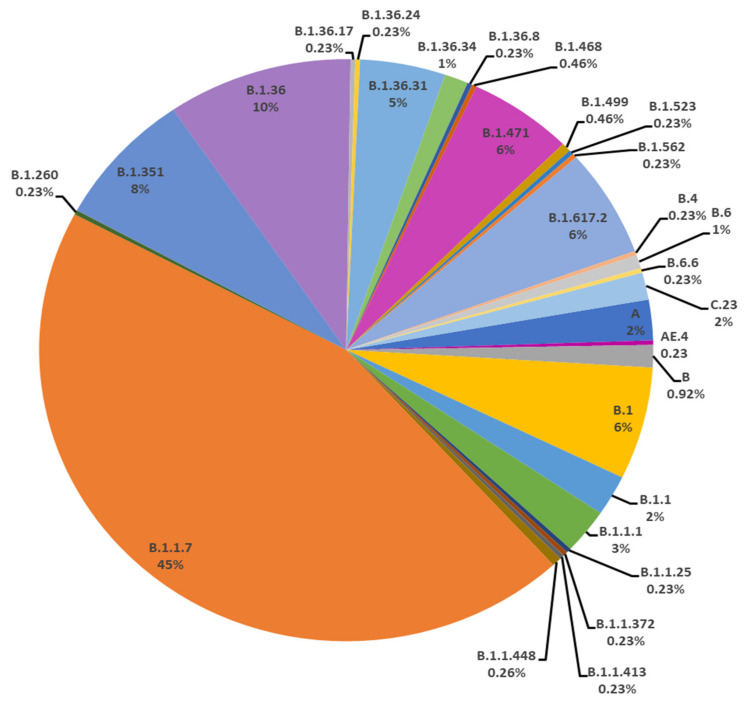
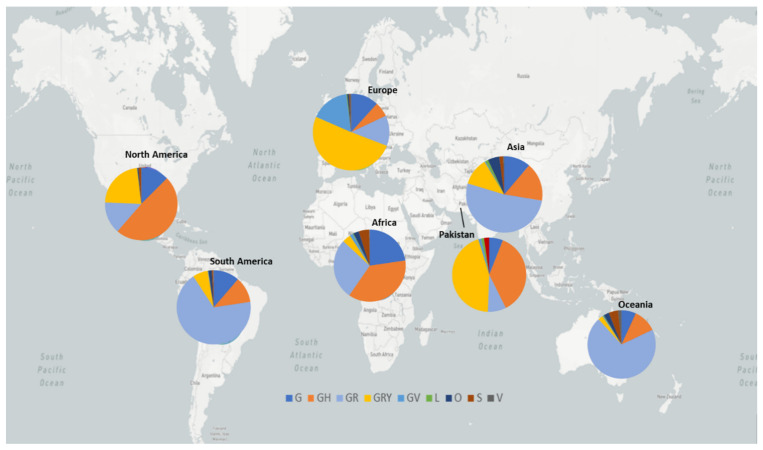
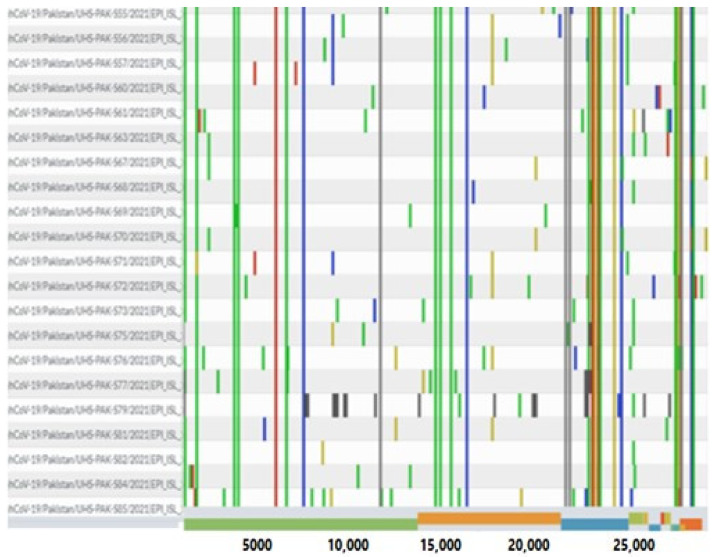
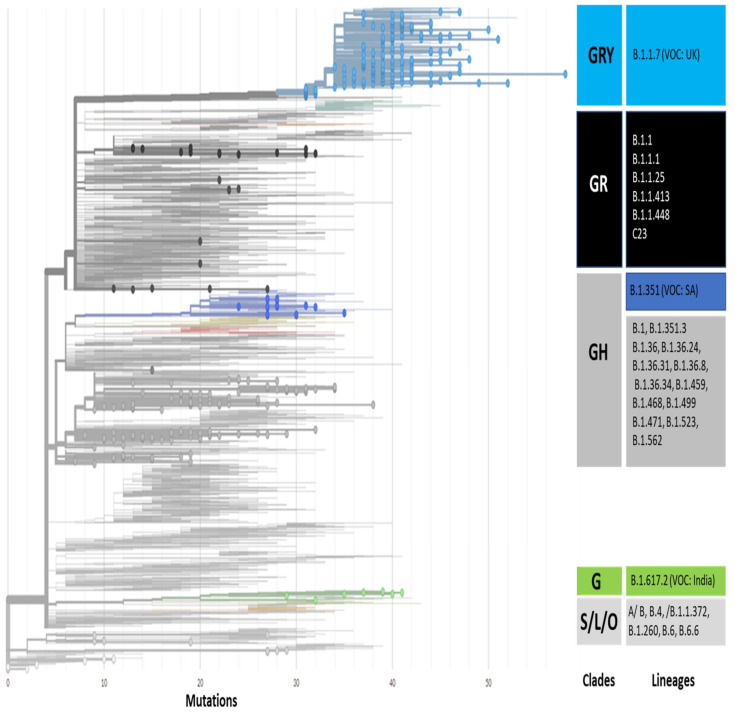
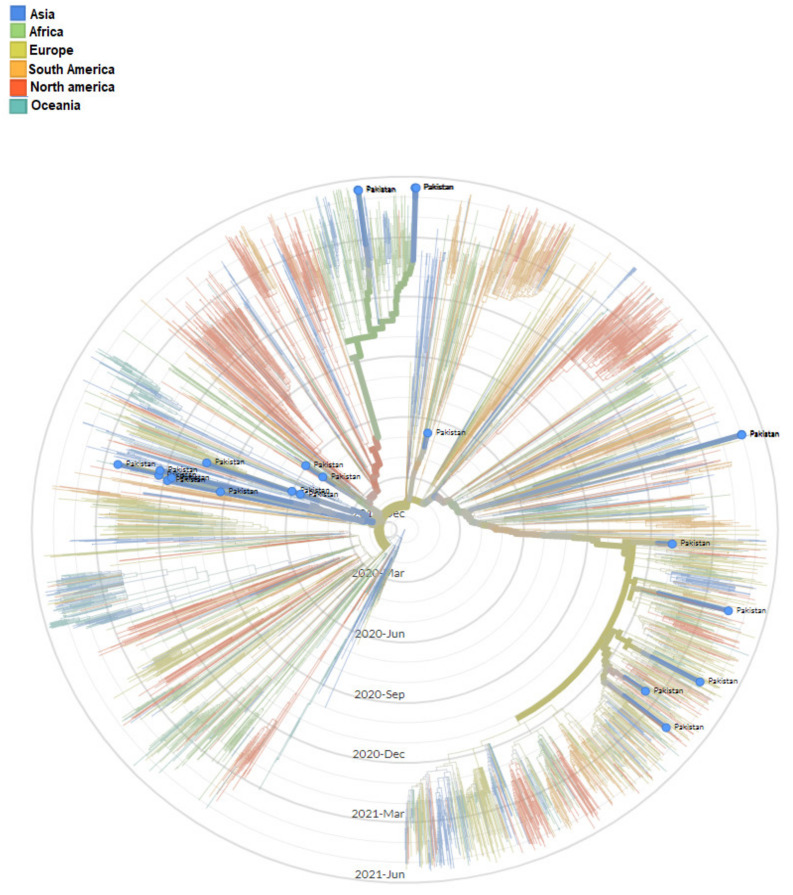
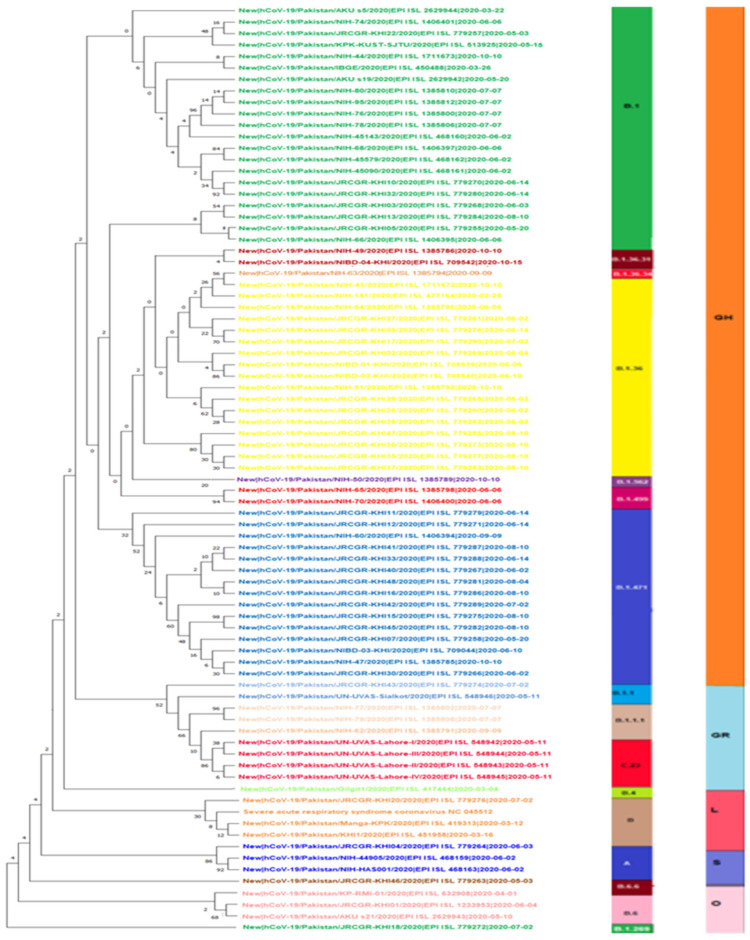
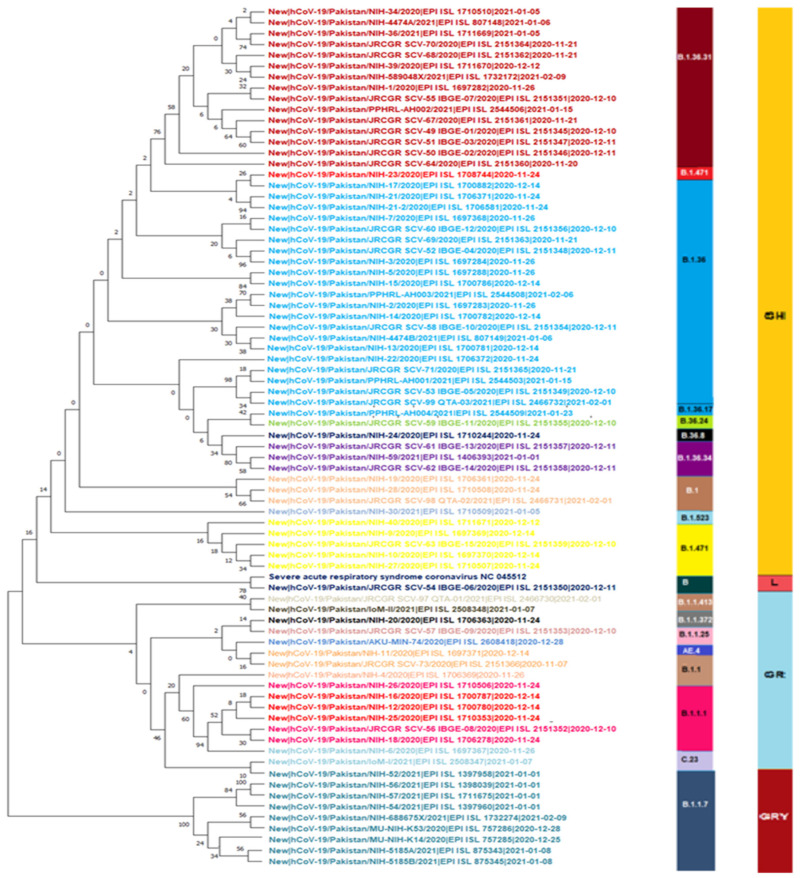
The present study aims to investigate the genomic variability and epidemiology of SARS-CoV-2 in Pakistan along with its role in the spread and severity of infection during the three waves of COVID-19. A total of 453 genomic sequences of Pakistani SARS-CoV-2 were retrieved from GISAID and subjected to MAFFT-based alignment and QC check which resulted in removal of 53 samples. The remaining 400 samples were subjected to Pangolin-based genomic lineage identification. And to infer our SARS-CoV-2 time-scaled and divergence phylogenetic trees, 3804 selected global reference sequences plus 400 Pakistani samples were used for the Nextstrain analysis with Wuhan/Hu-1/2019, as reference genome. Finally, maximum likelihood based phylogenetic tree was built by using the Nextstrain and coverage map was created by employing Nextclade. By using the amino acid substitutions, the maximum likelihood phylogenetic trees were developed for each wave, separately. Our results reveal the circulation of 29 lineages, belonging to following seven clades G, GH, GR, GRY, L, O, and S in the three waves. From first wave, 16 genomic lineages of SARS-CoV-2 were identified with B.1(24.7%), B.1.36(18.8%), and B.1.471(18.8%) as the most prevalent lineages respectively. The second wave data showed 18 lineages, 10 of which were overlapping with the first wave suggesting that those variants could not be contained during the first wave. In this wave, a new lineage, AE.4, was reported from Pakistan for the very first time in the world. However, B.1.36 (17.8%), B.1.36.31 (11.9%), B.1.1.7 (8.5%), and B.1.1.1 (5.9%) were the major lineages in second wave. Third wave data showed the presence of nine lineages with Alpha/B.1.1.7 (72.7%), Beta/B.1.351 (12.99%), and Delta/B.1.617.2 (10.39%) as the most predominant variants. It is suggested that these VOCs should be contained at the earliest in order to prevent any devastating outbreak of SARS-CoV-2 in the country.
本研究旨在调查巴基斯坦严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的基因组变异性和流行病学,以及其在2019冠状病毒病(COVID-19)三波疫情期间在感染传播和严重程度方面所起的作用。从全球共享流感数据倡议组织(GISAID)检索了总共453条巴基斯坦SARS-CoV-2基因组序列,并进行基于MAFFT的比对和质量控制检查,结果剔除了53个样本。其余400个样本进行基于穿山甲(Pangolin)的基因组谱系鉴定。为了推断我们的SARS-CoV-2时间尺度和分歧系统发育树,将3804条选定的全球参考序列加上400个巴基斯坦样本用于以武汉/胡-1/2019作为参考基因组的Nextstrain分析。最后,使用Nextstrain构建基于最大似然法的系统发育树,并使用Nextclade创建覆盖图。通过氨基酸替换,分别为每一波构建基于最大似然法的系统发育树。我们的结果显示,在三波疫情中共有29个谱系传播,分属于以下七个进化枝:G、GH、GR、GRY、L、O和S。在第一波疫情中,鉴定出16个SARS-CoV-2基因组谱系,其中B.1(24.7%)、B.1.36(18.8%)和B.1.471(18.8%)为最主要的谱系。第二波疫情数据显示有18个谱系,其中10个与第一波重叠,这表明这些变异株在第一波疫情期间未能得到控制。在这一波疫情中,一个新的谱系AE.4首次在世界范围内从巴基斯坦被报道。然而,B.1.36(17.8%)、B.1.36.31(11.9%)、B.1.1.7(8.5%)和B.1.1.1(5.9%)是第二波疫情中的主要谱系。第三波疫情数据显示有9个谱系,其中Alpha/B.1.1.7(72.7%)、Beta/B.1.351(12.99%)和Delta/B.1.617.2(10.39%)为最主要的变异株。建议尽早控制这些变异株,以防止该国出现任何毁灭性的SARS-CoV-2疫情爆发。