Institute of Computer Science, Martin Luther University Halle-Wittenberg, Halle, Germany.
Institute of Plant Genetics, Leibniz Universität Hannover, Hannover, Germany.
BMC Genomics. 2021 Dec 29;22(1):914. doi: 10.1186/s12864-021-08210-z.
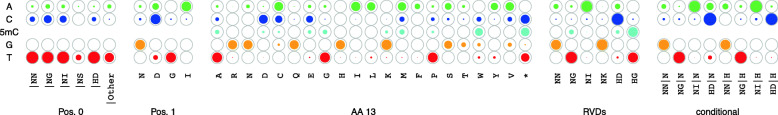
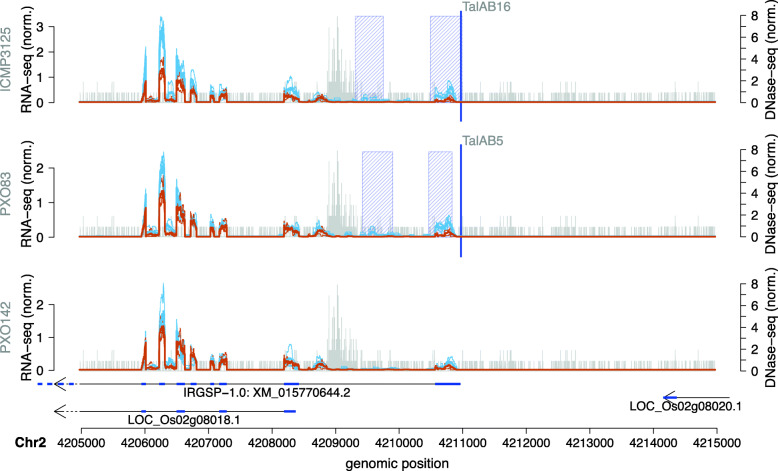
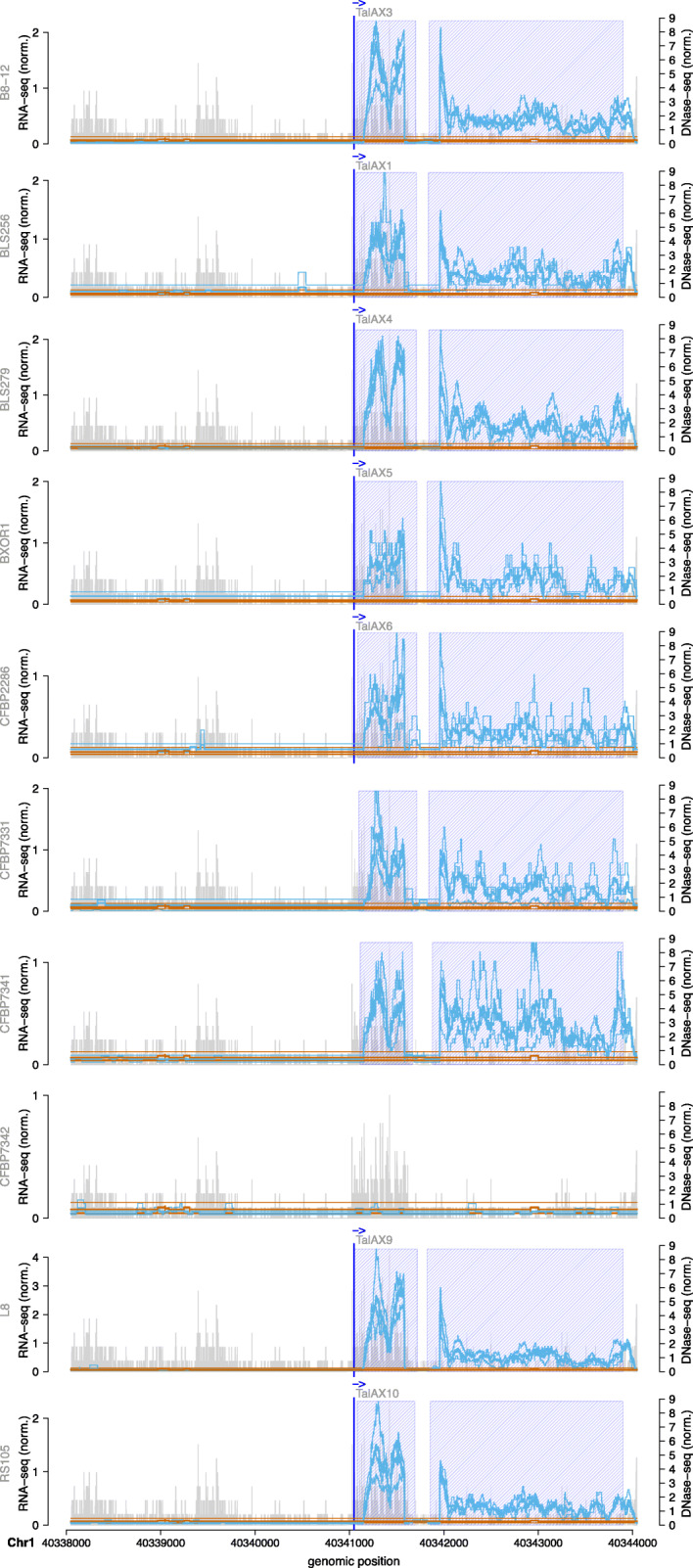
The yield of many crop plants can be substantially reduced by plant-pathogenic Xanthomonas bacteria. The infection strategy of many Xanthomonas strains is based on transcription activator-like effectors (TALEs), which are secreted into the host cells and act as transcriptional activators of plant genes that are beneficial for the bacteria.The modular DNA binding domain of TALEs contains tandem repeats, each comprising two hyper-variable amino acids. These repeat-variable diresidues (RVDs) bind to their target box and determine the specificity of a TALE.All available tools for the prediction of TALE targets within the host plant suffer from many false positives. In this paper we propose a strategy to improve prediction accuracy by considering the epigenetic state of the host plant genome in the region of the target box.
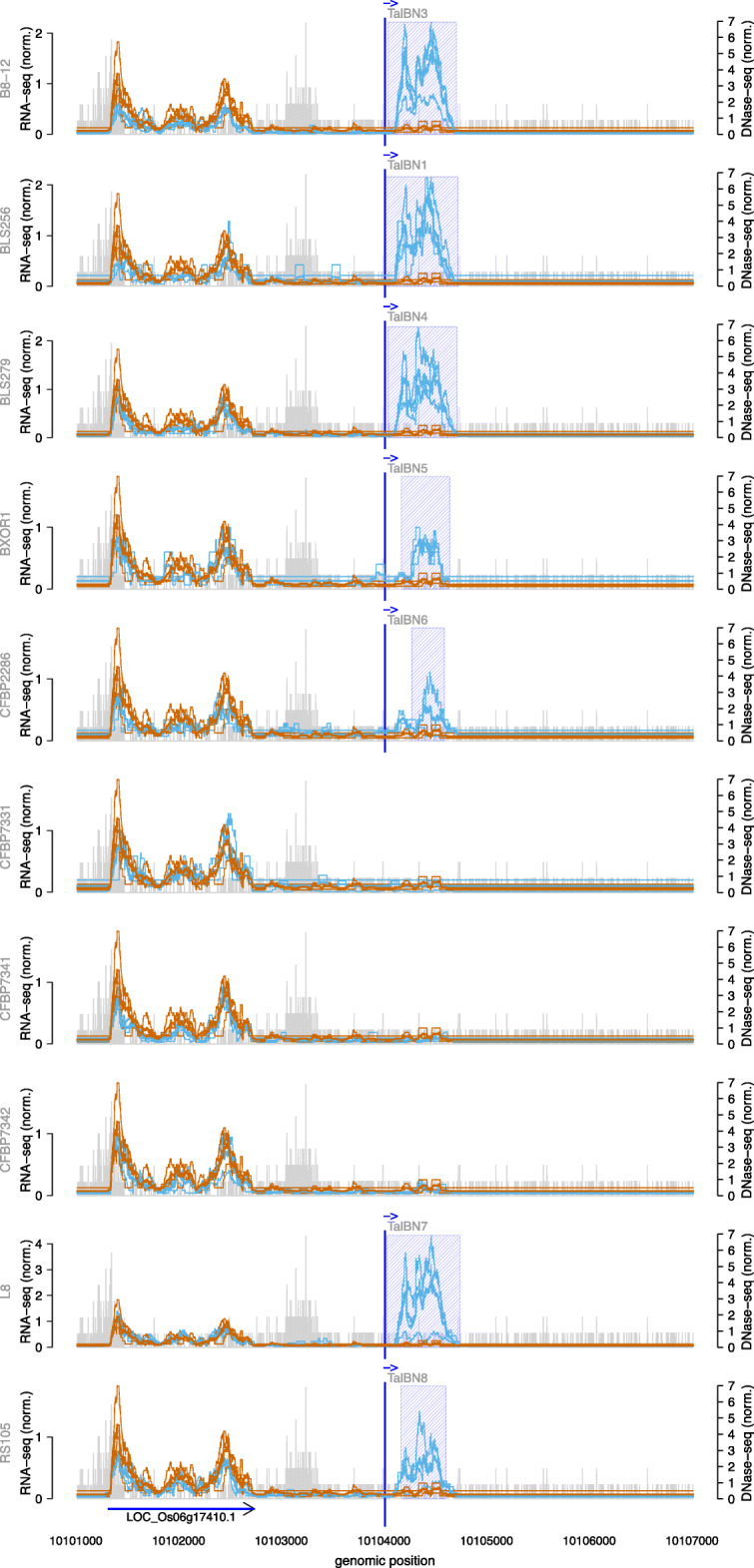
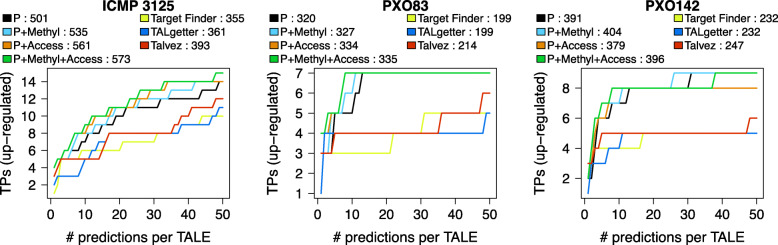
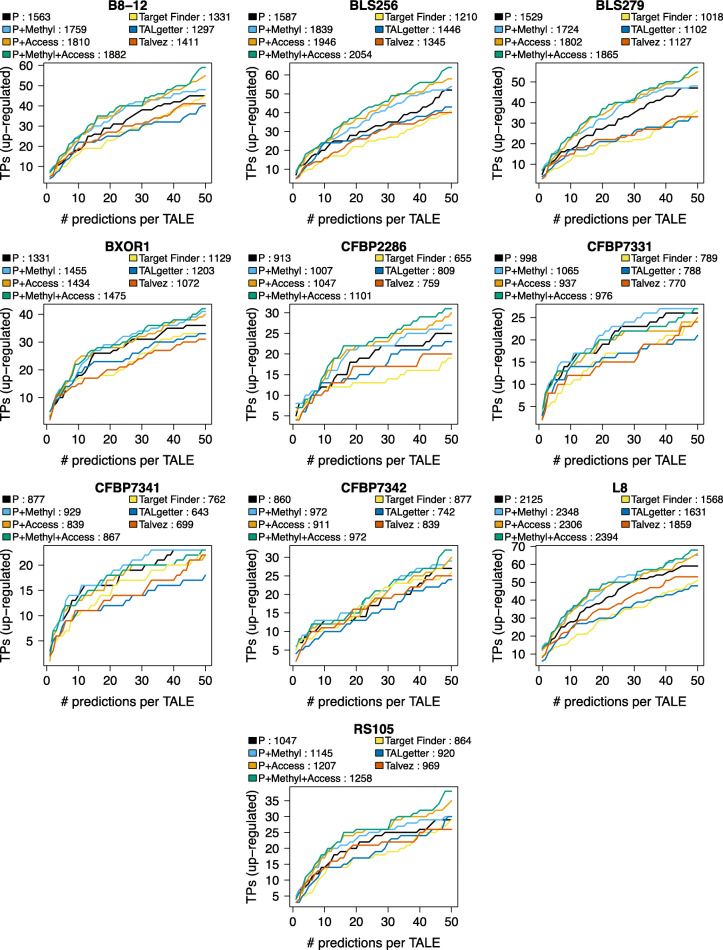
To this end, we extend our previously published tool PrediTALE by considering two epigenetic features: (i) chromatin accessibility of potentially bound regions and (ii) DNA methylation of cytosines within target boxes. Here, we determine the epigenetic features from publicly available DNase-seq, ATAC-seq, and WGBS data in rice.We benchmark the utility of both epigenetic features separately and in combination, deriving ground-truth from RNA-seq data of infections studies in rice. We find an improvement for each individual epigenetic feature, but especially the combination of both.Having established an advantage in TALE target predicting considering epigenetic features, we use these data for promoterome and genome-wide scans by our new tool EpiTALE, leading to several novel putative virulence targets.
Our results suggest that it would be worthwhile to collect condition-specific chromatin accessibility data and methylation information when studying putative virulence targets of Xanthomonas TALEs.
植物病原黄单胞菌会显著降低许多作物的产量。许多黄单胞菌菌株的感染策略基于转录激活子样效应物(TALEs),这些效应物被分泌到宿主细胞中,并作为有利于细菌的植物基因的转录激活子发挥作用。TALEs 的模块化 DNA 结合域包含串联重复序列,每个重复序列由两个超变氨基酸组成。这些重复可变二氨基酸(RVD)与它们的靶盒结合,决定了 TALE 的特异性。目前所有用于预测宿主植物中 TALE 靶标的工具都存在许多假阳性。在本文中,我们提出了一种通过考虑靶盒区域中宿主植物基因组的表观遗传状态来提高预测准确性的策略。
为此,我们通过考虑两个表观遗传特征来扩展我们之前发表的工具 PrediTALE:(i)潜在结合区域的染色质可及性和(ii)靶盒内胞嘧啶的 DNA 甲基化。在这里,我们从水稻中公开的 DNase-seq、ATAC-seq 和 WGBS 数据中确定了这些表观遗传特征。我们分别和组合使用这两种表观遗传特征进行基准测试,从水稻感染研究的 RNA-seq 数据中得出真实情况。我们发现每个单独的表观遗传特征都有所改进,但特别是两者的组合。在考虑表观遗传特征的 TALE 靶标预测方面取得了优势之后,我们使用这些数据通过我们的新工具 EpiTALE 进行启动子组和全基因组扫描,从而发现了几个新的潜在毒力靶标。
我们的结果表明,在研究黄单胞菌 TALEs 的潜在毒力靶标时,收集特定条件下的染色质可及性数据和甲基化信息将是值得的。