Universidade Federal do Rio de Janeiro, Instituto de Biologia, Departamento de Genética, Laboratório de Virologia Molecular, Rio de Janeiro, RJ, Brasil.
Universidade Federal do Rio de Janeiro, Instituto de Biologia, Departamento de Genética, Laboratório de Diversidade e Doenças Virais, Rio de Janeiro, RJ, Brasil.
Mem Inst Oswaldo Cruz. 2022 Jan 10;116:e210176. doi: 10.1590/0074-02760210176. eCollection 2022.
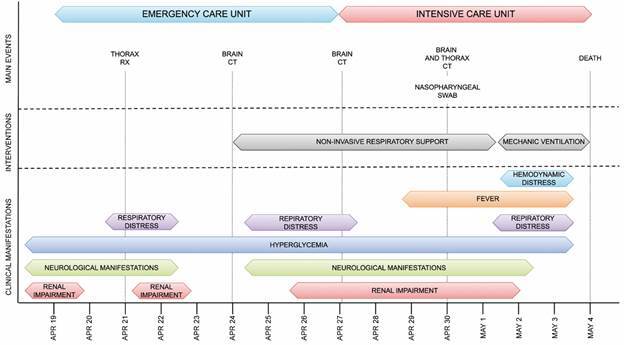
During routine Coronavirus disease 2019 (COVID-19) diagnosis, an unusually high viral load was detected by reverse transcription real-time polymerase chain reaction (RT-qPCR) in a nasopharyngeal swab sample collected from a patient with respiratory and neurological symptoms who rapidly succumbed to the disease. Therefore we sought to characterise the infection.
We aimed to determine and characterise the etiological agent responsible for the poor outcome.
Classical virological methods, such as plaque assay and plaque reduction neutralisation test combined with amplicon-based sequencing, as well as a viral metagenomic approach, were performed to characterise the etiological agents of the infection.
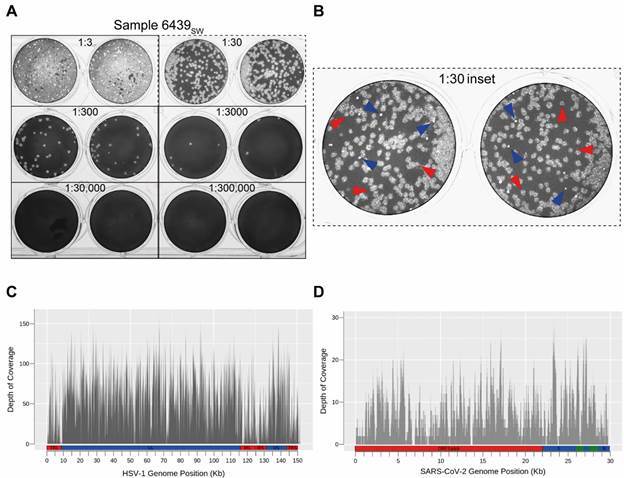
Plaque assay revealed two distinct plaque phenotypes, suggesting either the presence of two severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) strains or a productive coinfection of two different species of virus. Amplicon-based sequencing did not support the presence of any SARS-CoV-2 genetic variants that would explain the high viral load and suggested the presence of a single SARS-CoV-2 strain. Nonetheless, the viral metagenomic analysis revealed that Coronaviridae and Herpesviridae were the predominant virus families within the sample. This finding was confirmed by a plaque reduction neutralisation test and PCR.
We characterised a productive coinfection of SARS-CoV-2 and Herpes simplex virus 1 (HSV-1) in a patient with severe symptoms that succumbed to the disease. Although we cannot establish the causal relationship between the coinfection and the severity of the clinical case, this work serves as a warning for future studies focused on the interplay between SARS-CoV-2 and HSV-1 coinfection and COVID-19 severity.
在常规的 2019 年冠状病毒病(COVID-19)诊断过程中,从一名患有呼吸和神经系统症状并迅速死于该病的患者的鼻咽拭子样本中,通过逆转录实时聚合酶链反应(RT-qPCR)检测到异常高的病毒载量。因此,我们试图描述这种感染。
我们旨在确定和描述导致不良结果的病原体。
采用经典病毒学方法,如噬斑测定和噬斑减少中和试验结合基于扩增子的测序,以及病毒宏基因组学方法,来描述感染的病原体。
噬斑测定显示出两种不同的噬斑表型,这表明存在两种严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)株,或者是两种不同病毒的有效合并感染。基于扩增子的测序不支持存在任何可解释高病毒载量的 SARS-CoV-2 遗传变异体,并表明存在单一 SARS-CoV-2 株。尽管如此,病毒宏基因组分析显示冠状病毒科和疱疹病毒科是样本中主要的病毒科。这一发现通过噬斑减少中和试验和 PCR 得到了证实。
我们在一名患有严重症状并死于该病的患者中描述了 SARS-CoV-2 和单纯疱疹病毒 1(HSV-1)的有效合并感染。虽然我们不能确定合并感染与临床病例严重程度之间的因果关系,但这项工作为未来研究 SARS-CoV-2 和 HSV-1 合并感染与 COVID-19 严重程度之间的相互作用提供了警示。