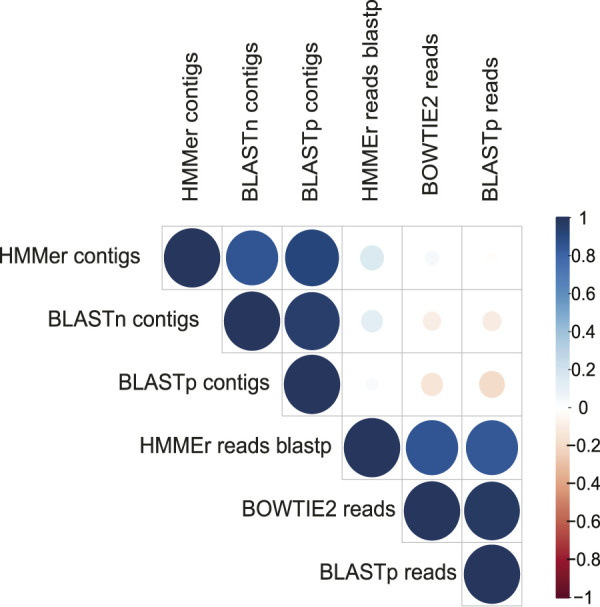
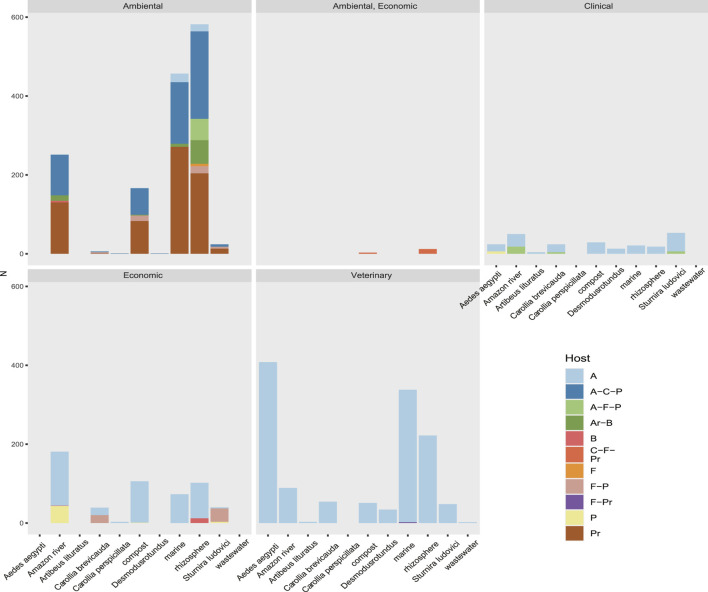
Mazur Fernando G, Morinisi Leandro M, Martins Junior Olímpio, Guerra Pedro Pontes Bueno, Freire Caio C M
Department Genetics and Evolution, UFSCar-Federal University of São Carlos, São Carlos, Brazil.
Front Genet. 2022 Jan 21;12:722857. doi: 10.3389/fgene.2021.722857. eCollection 2021.
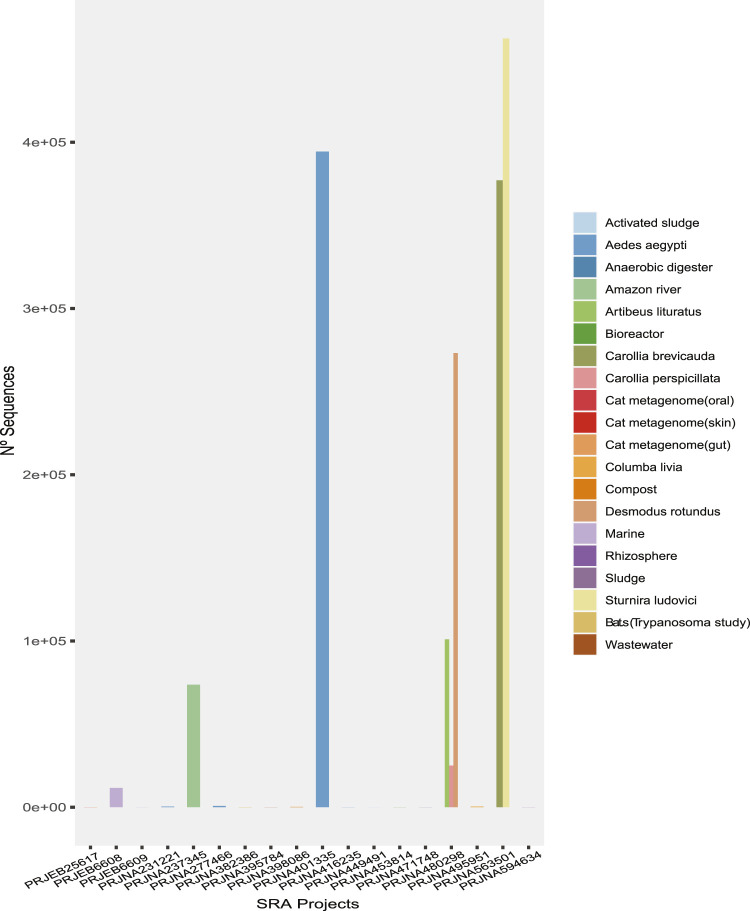
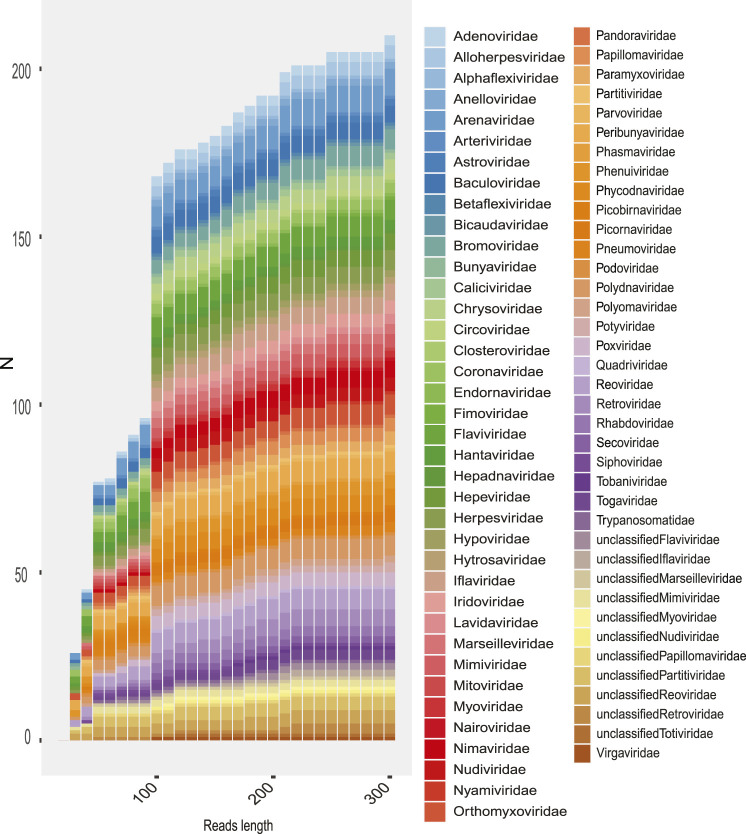
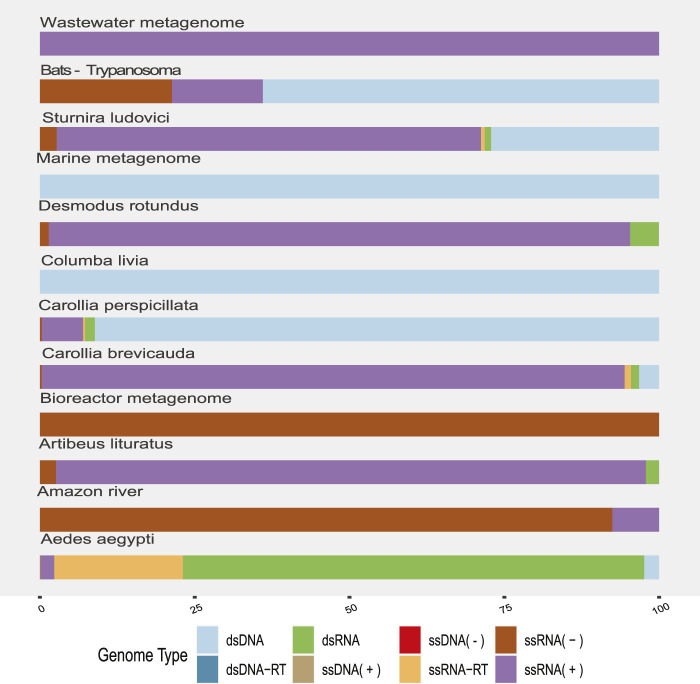
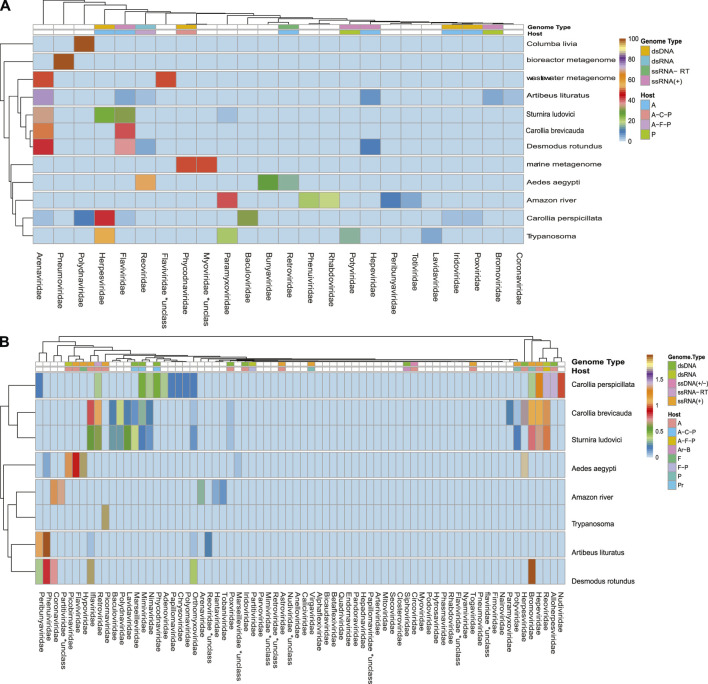
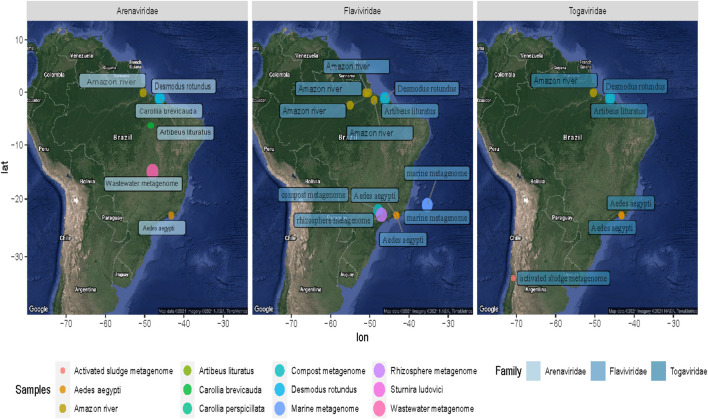
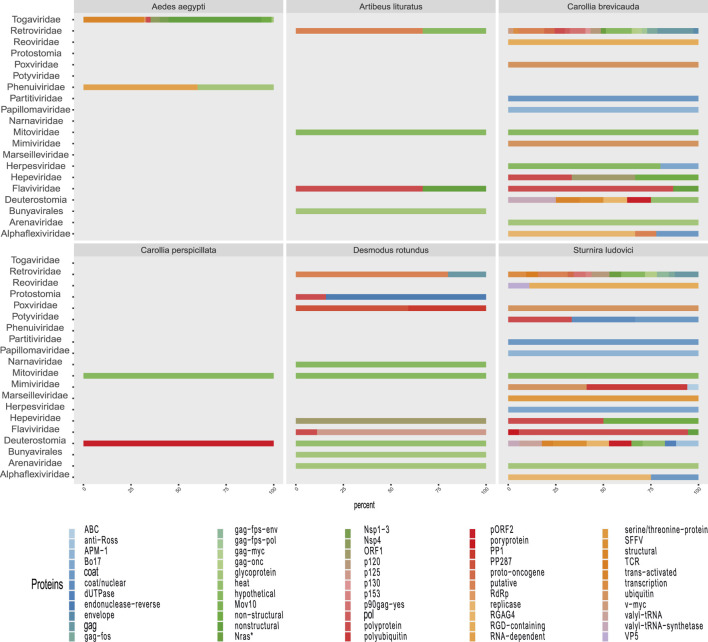
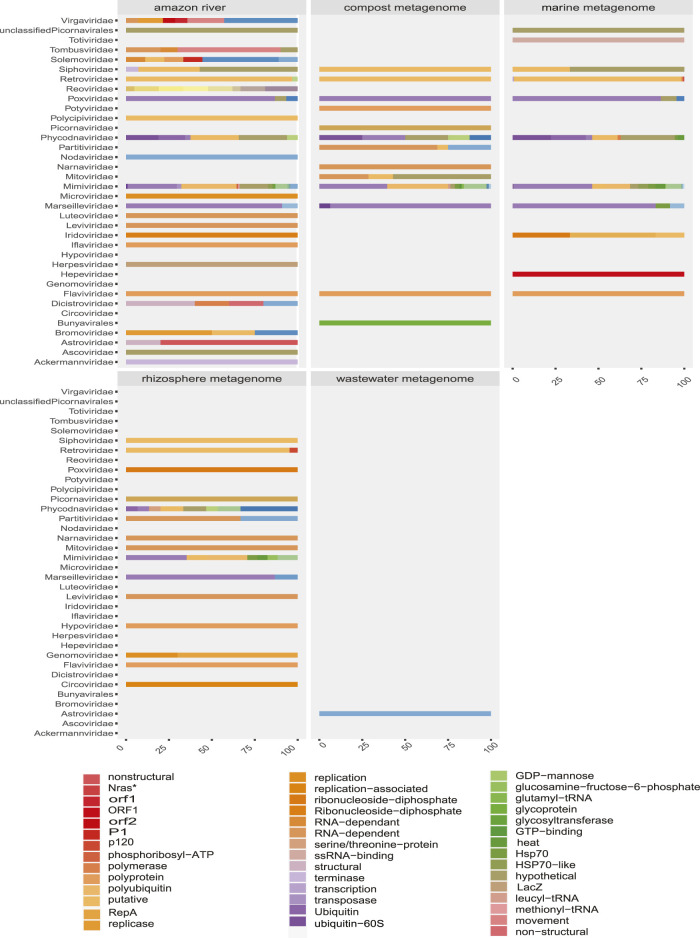
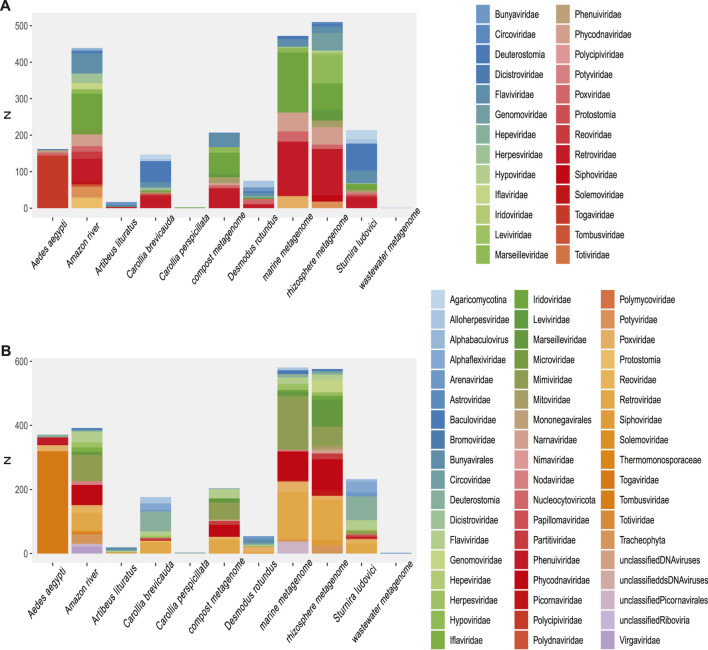
The South American continent presents a great diversity of biomes, whose ecosystems are constantly threatened by the expansion of human activity. The emergence and re-emergence of viral populations with impact on the human population and ecosystem have shown increases in the last decades. In deference to the growing accumulation of genomic data, we explore the potential of South American-related public databases to detect signals that contribute to virosphere research. Therefore, our study aims to investigate public databases with emphasis on the surveillance of viruses with medical and ecological relevance. Herein, we profiled 120 "" metagenomes from 19 independent projects from the last decade. In a coarse view, our analyses identified only 0.38% of the total number of sequences from viruses, showing a higher proportion of RNA viruses. The metagenomes with the most important viral sequences in the analyzed environmental models were 1) aquatic samples from the Amazon River, 2) sewage from Brasilia, and 3) soil from the state of São Paulo, while the models of animal transmission were detected in mosquitoes from Rio Janeiro and Bats from Amazonia. Also, the classification of viral signals into operational taxonomic units (OTUs) (family) allowed us to infer from metadata a probable host range in the virome detected in each sample analyzed. Further, several motifs and viral sequences are related to specific viruses with emergence potential from , , and families. In this context, the exploration of public databases allowed us to evaluate the scope and informative capacity of sequences from third-party public databases and to detect signals related to viruses of clinical or environmental importance, which allowed us to infer traits associated with probable transmission routes or signals of ecological disequilibrium. The evaluation of our results showed that in most cases the size and type of the reference database, the percentage of guanine-cytosine (GC), and the length of the query sequences greatly influence the taxonomic classification of the sequences. In sum, our findings describe how the exploration of public genomic data can be exploited as an approach for epidemiological surveillance and the understanding of the virosphere.
南美洲大陆呈现出多种多样的生物群落,其生态系统不断受到人类活动扩张的威胁。在过去几十年中,对人类种群和生态系统有影响的病毒种群的出现和再次出现呈上升趋势。鉴于基因组数据的不断积累,我们探索了与南美洲相关的公共数据库在检测有助于病毒圈研究的信号方面的潜力。因此,我们的研究旨在调查公共数据库,重点关注具有医学和生态相关性的病毒监测。在此,我们剖析了过去十年中来自19个独立项目的120个宏基因组。粗略来看,我们的分析仅识别出病毒序列总数的0.38%,显示出RNA病毒的比例更高。在所分析的环境模型中,具有最重要病毒序列的宏基因组分别是:1)亚马逊河的水生样本,2)巴西利亚的污水,以及3)圣保罗州的土壤,而在动物传播模型中,在里约热内卢的蚊子和亚马逊地区的蝙蝠中检测到相关病毒。此外,将病毒信号分类为操作分类单元(OTU)(科)使我们能够从元数据中推断出在每个分析样本中检测到的病毒群落中可能的宿主范围。此外,几个基序和病毒序列与来自 、 和 科的具有出现潜力的特定病毒相关。在这种背景下,对公共数据库的探索使我们能够评估来自第三方公共数据库的序列的范围和信息容量,并检测与具有临床或环境重要性的病毒相关的信号,这使我们能够推断与可能的传播途径或生态失衡信号相关的特征。对我们结果的评估表明,在大多数情况下,参考数据库的大小和类型、鸟嘌呤 - 胞嘧啶(GC)百分比以及查询序列的长度对序列的分类学分类有很大影响。总之,我们的研究结果描述了如何利用公共基因组数据探索作为一种流行病学监测方法以及对病毒圈的理解。