Center for Translational Data Science, The University of Chicago, Chicago, IL, 60615, USA.
Department of Medicine, The University of Chicago, Chicago, IL, 60637, USA.
Sci Rep. 2022 Feb 14;12(1):2427. doi: 10.1038/s41598-022-06449-4.
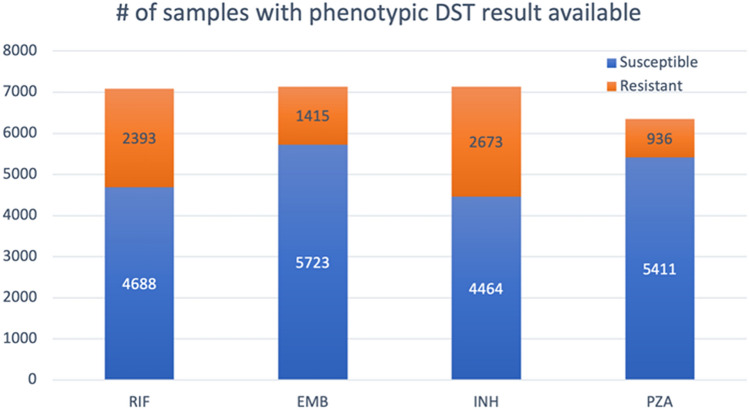
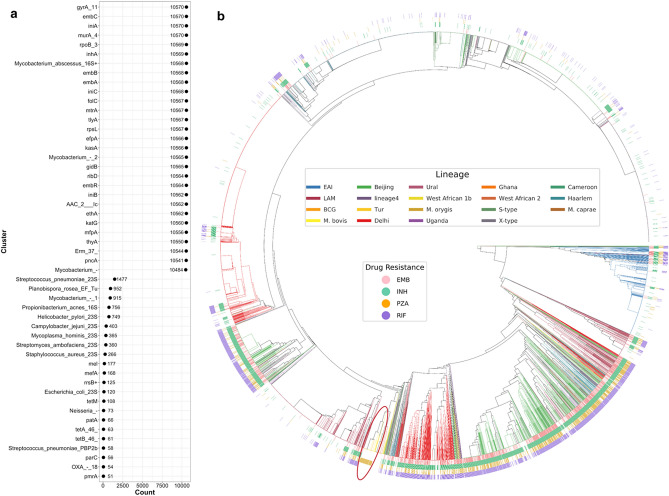
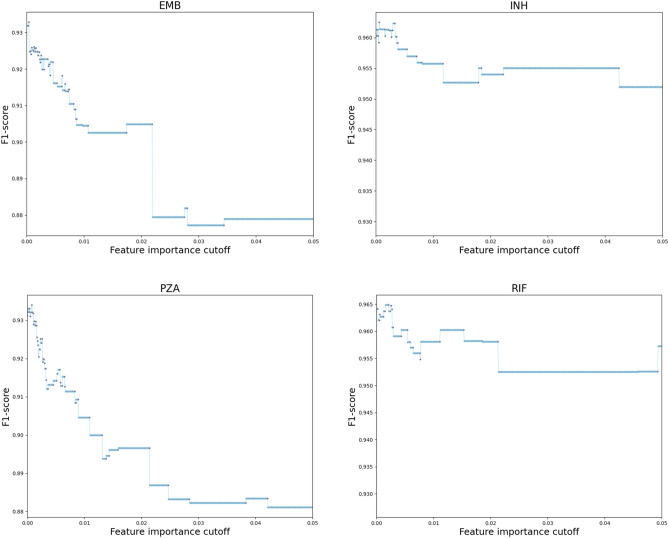
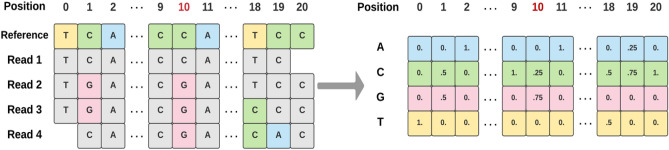
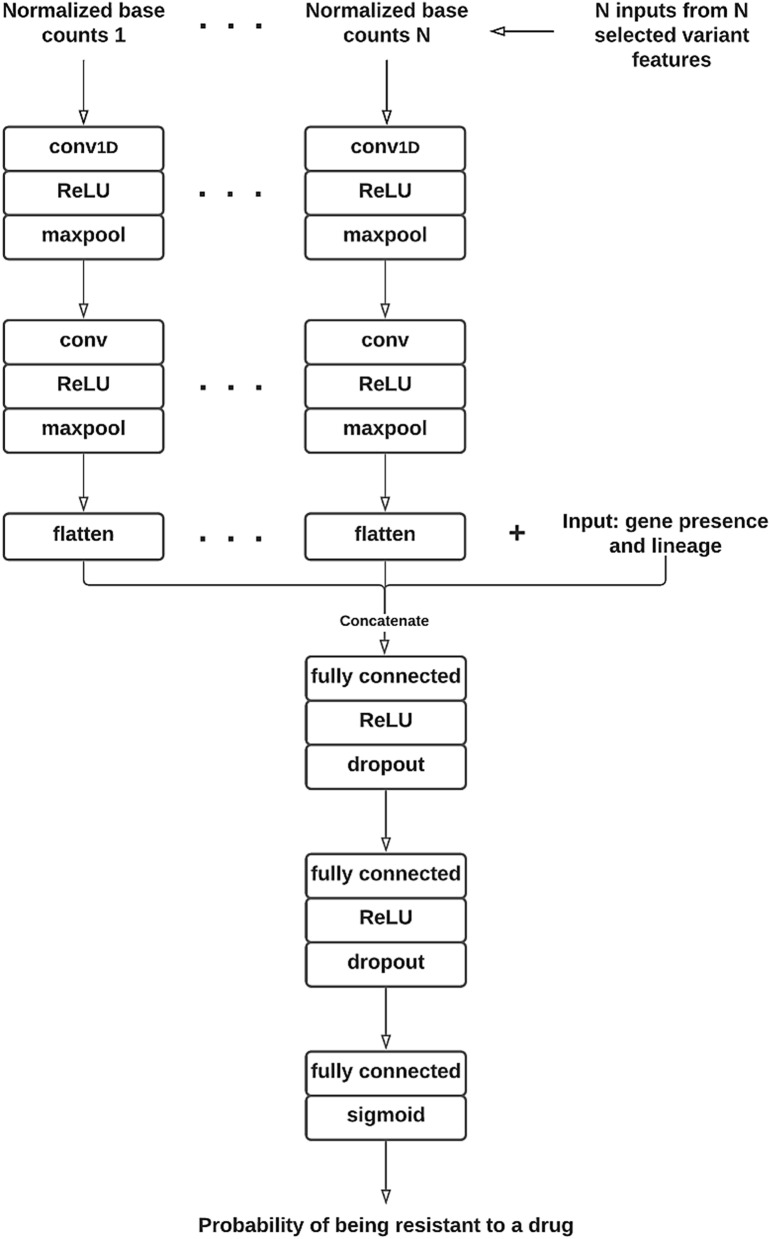
Effective and timely antibiotic treatment depends on accurate and rapid in silico antimicrobial-resistant (AMR) predictions. Existing statistical rule-based Mycobacterium tuberculosis (MTB) drug resistance prediction methods using bacterial genomic sequencing data often achieve varying results: high accuracy on some antibiotics but relatively low accuracy on others. Traditional machine learning (ML) approaches have been applied to classify drug resistance for MTB and have shown more stable performance. However, there is no study that uses deep learning architecture like Convolutional Neural Network (CNN) on a large and diverse cohort of MTB samples for AMR prediction. We developed 24 binary classifiers of MTB drug resistance status across eight anti-MTB drugs and three different ML algorithms: logistic regression, random forest and 1D CNN using a training dataset of 10,575 MTB isolates collected from 16 countries across six continents, where an extended pan-genome reference was used for detecting genetic features. Our 1D CNN architecture was designed to integrate both sequential and non-sequential features. In terms of F1-scores, 1D CNN models are our best classifiers that are also more accurate and stable than the state-of-the-art rule-based tool Mykrobe predictor (81.1 to 93.8%, 93.7 to 96.2%, 93.1 to 94.8%, 95.9 to 97.2% and 97.1 to 98.2% for ethambutol, rifampicin, pyrazinamide, isoniazid and ofloxacin respectively). We applied filter-based feature selection to find AMR relevant features. All selected variant features are AMR-related ones in CARD database. 78.8% of them are also in the catalogue of MTB mutations that were recently identified as drug resistance-associated ones by WHO. To facilitate ML model development for AMR prediction, we packaged every step into an automated pipeline and shared the source code at https://github.com/KuangXY3/MTB-AMR-classification-CNN .
有效的、及时的抗生素治疗取决于准确且快速的计算抗菌药物耐药性(AMR)预测。现有的基于统计规则的结核分枝杆菌(MTB)耐药预测方法使用细菌基因组测序数据,往往会得到不同的结果:对某些抗生素的准确性较高,但对其他抗生素的准确性相对较低。传统的机器学习(ML)方法已被应用于 MTB 的耐药性分类,并表现出更稳定的性能。然而,目前还没有研究使用卷积神经网络(CNN)等深度学习架构对来自六大洲 16 个国家的大量、多样化的 MTB 样本进行 AMR 预测。我们使用来自六大洲 16 个国家的 10575 株 MTB 分离株的训练数据集,开发了 24 种针对八种抗 MTB 药物的 MTB 耐药状态的二元分类器,并使用三种不同的 ML 算法:逻辑回归、随机森林和 1DCNN。其中,扩展的泛基因组参考用于检测遗传特征。我们的 1DCNN 架构旨在整合顺序和非顺序特征。在 F1 分数方面,1DCNN 模型是我们最好的分类器,比最先进的基于规则的工具 Mykrobe predictor(分别为 81.1%至 93.8%、93.7%至 96.2%、93.1%至 94.8%、95.9%至 97.2%和 97.1%至 98.2%)更准确和稳定,分别用于乙胺丁醇、利福平、吡嗪酰胺、异烟肼和氧氟沙星。我们应用基于滤波器的特征选择来发现 AMR 相关特征。所有选定的变体特征在 CARD 数据库中均与 AMR 相关。其中 78.8%也在世界卫生组织最近确定的与耐药性相关的 MTB 突变目录中。为了方便 AMR 预测的 ML 模型开发,我们将每个步骤打包到一个自动化管道中,并在 https://github.com/KuangXY3/MTB-AMR-classification-CNN 上共享源代码。