Sekula Michael, Gaskins Jeremy, Datta Susmita
Department of Bioinformatics and Biostatistics, University of Louisville, Louisville, KY, United States.
Department of Biostatistics, University of Florida, Gainesville, FL, United States.
Front Genet. 2022 Feb 4;12:810816. doi: 10.3389/fgene.2021.810816. eCollection 2021.
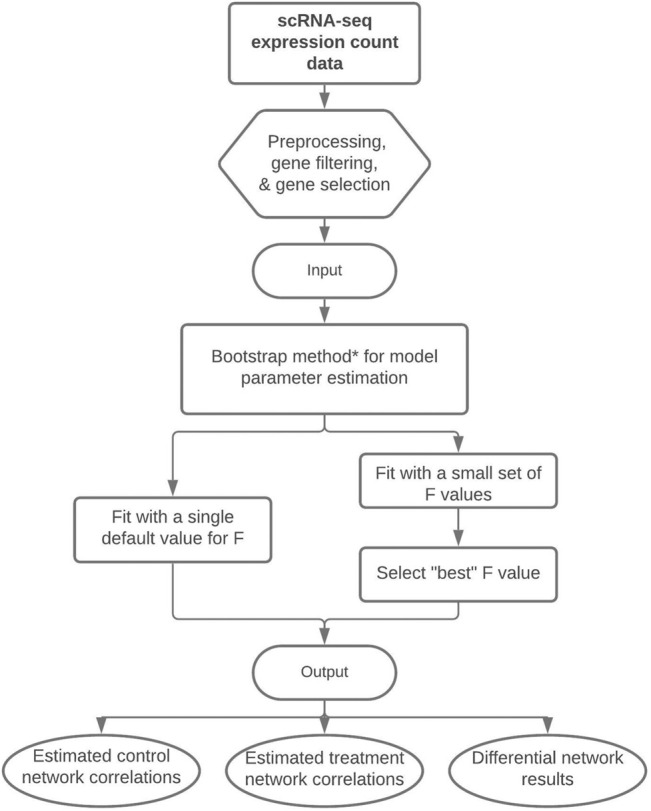
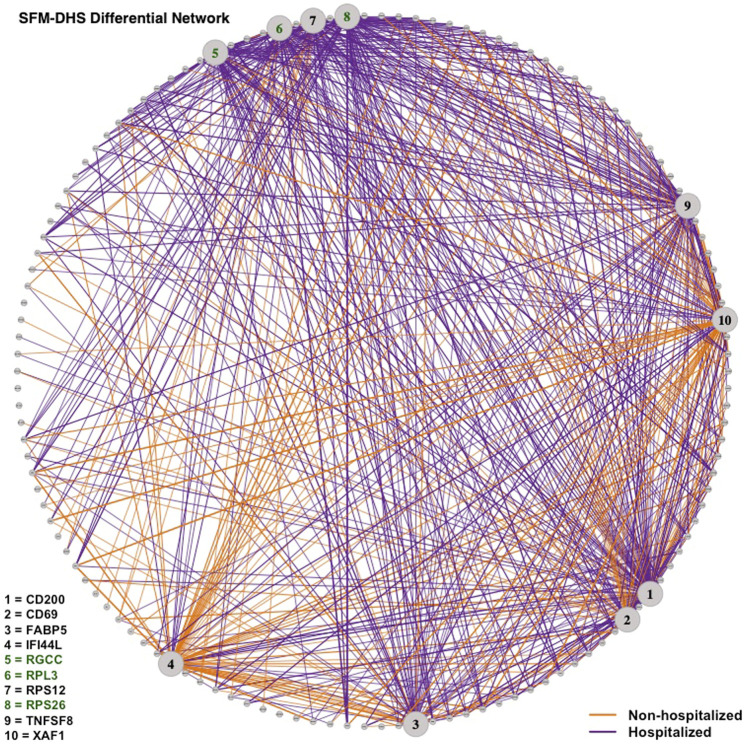
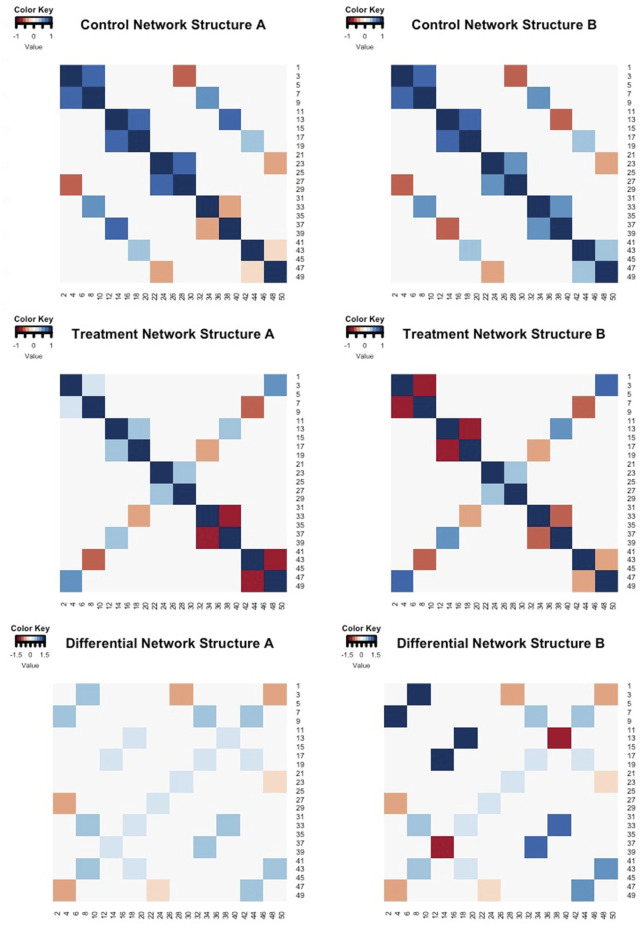
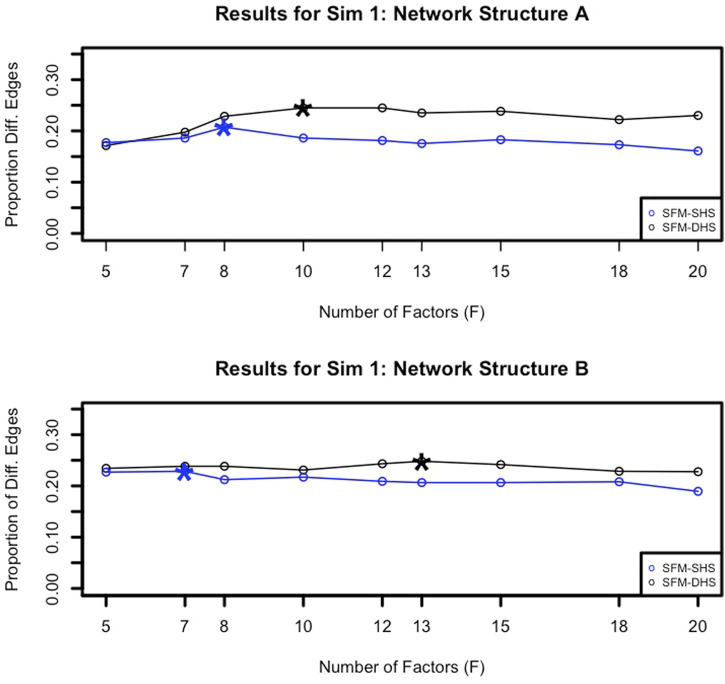
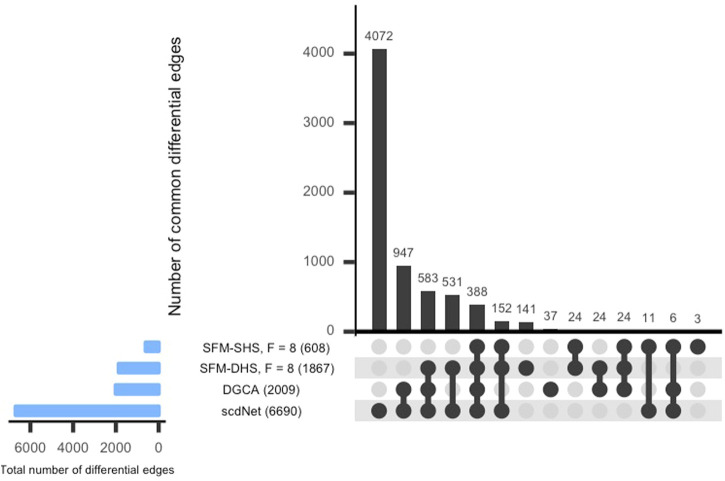
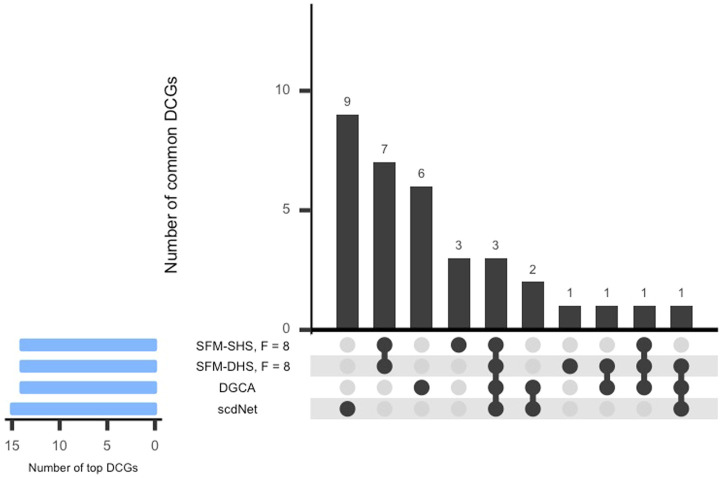
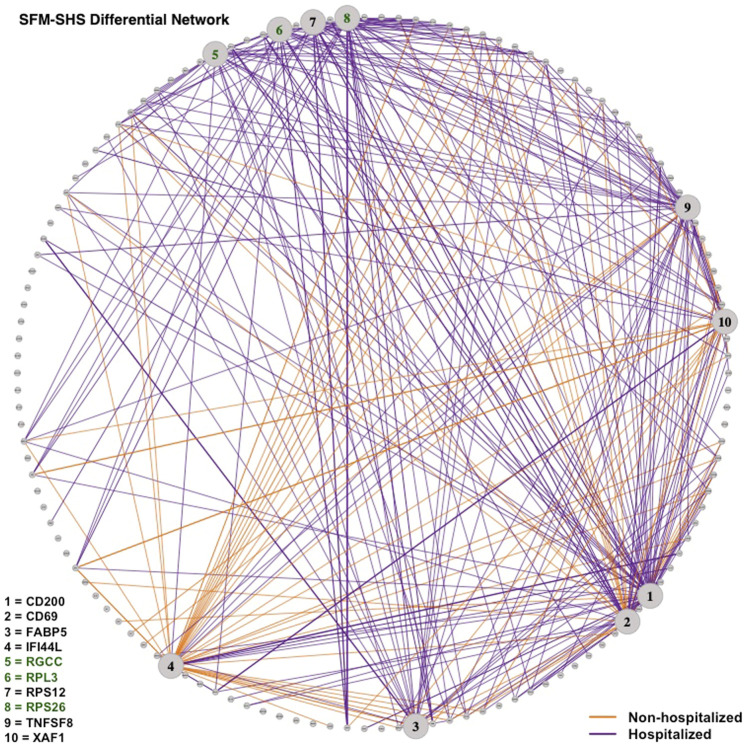
Differential network analysis plays an important role in learning how gene interactions change under different biological conditions, and the high resolution of single-cell RNA (scRNA-seq) sequencing provides new opportunities to explore these changing gene-gene interactions. Here, we present a sparse hierarchical Bayesian factor model to identify differences across network structures from different biological conditions in scRNA-seq data. Our methodology utilizes latent factors to impact gene expression values for each cell to help account for zero-inflation, increased cell-to-cell variability, and overdispersion that are unique characteristics of scRNA-seq data. Condition-dependent parameters determine which latent factors are activated in a gene, which allows for not only the calculation of gene-gene co-expression within each group but also the calculation of the co-expression differences between groups. We highlight our methodology's performance in detecting differential gene-gene associations across groups by analyzing simulated datasets and a SARS-CoV-2 case study dataset.
差异网络分析在了解基因相互作用如何在不同生物学条件下变化方面发挥着重要作用,而单细胞RNA(scRNA-seq)测序的高分辨率为探索这些不断变化的基因-基因相互作用提供了新机会。在此,我们提出一种稀疏分层贝叶斯因子模型,以识别scRNA-seq数据中不同生物学条件下网络结构的差异。我们的方法利用潜在因子来影响每个细胞的基因表达值,以帮助解释零膨胀、细胞间变异性增加和过度离散等scRNA-seq数据的独特特征。依赖于条件的参数决定了哪些潜在因子在一个基因中被激活,这不仅允许计算每组内的基因-基因共表达,还允许计算组间的共表达差异。我们通过分析模拟数据集和一个SARS-CoV-2案例研究数据集,突出了我们的方法在检测跨组差异基因-基因关联方面的性能。