San Diego Jewish Academy, San Diego, 92130, CA, USA.
California Institute of Technology, Pasadena, CA, 91125, USA.
J Comput Aided Mol Des. 2022 Feb;36(2):87-99. doi: 10.1007/s10822-021-00433-2. Epub 2022 Feb 24.
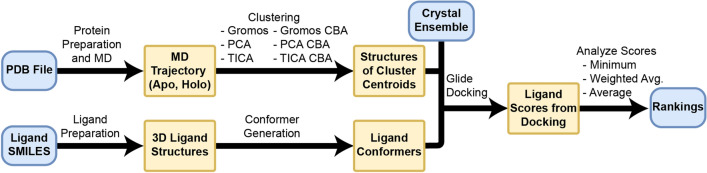
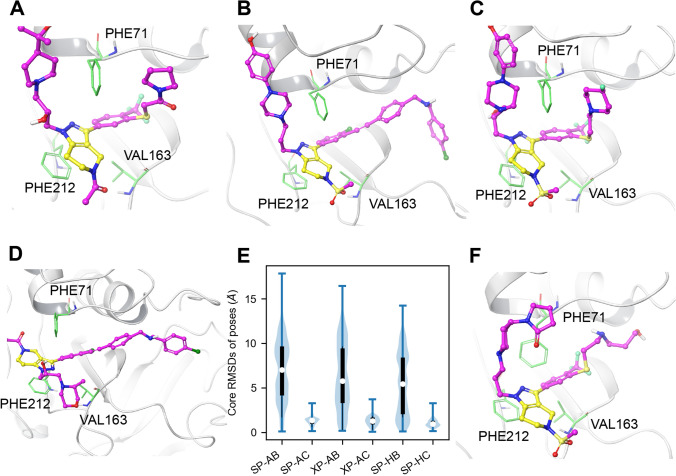
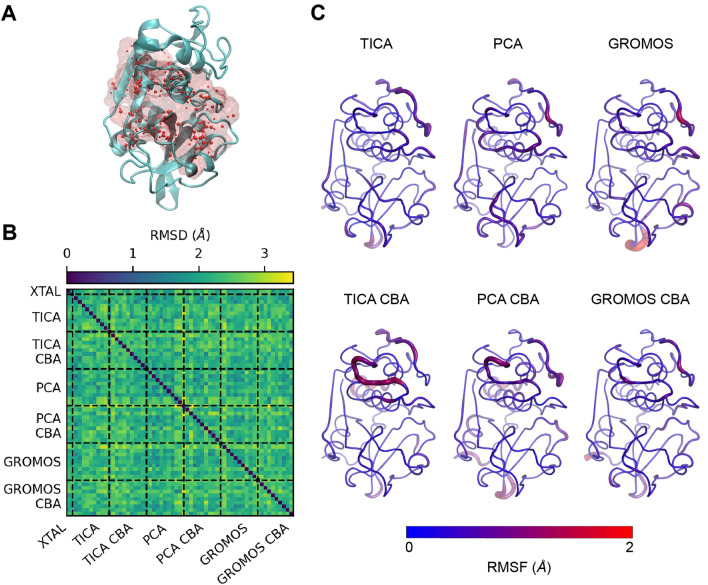
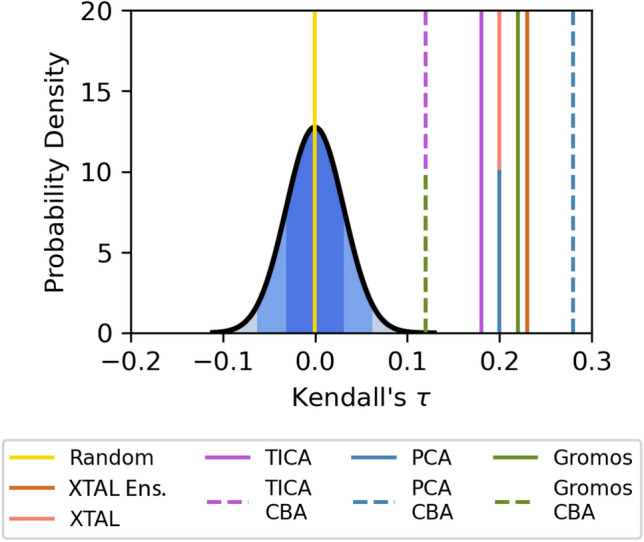
The discovery of new drugs is a time consuming and expensive process. Methods such as virtual screening, which can filter out ineffective compounds from drug libraries prior to expensive experimental study, have become popular research topics. As the computational drug discovery community has grown, in order to benchmark the various advances in methodology, organizations such as the Drug Design Data Resource have begun hosting blinded grand challenges seeking to identify the best methods for ligand pose-prediction, ligand affinity ranking, and free energy calculations. Such open challenges offer a unique opportunity for researchers to partner with junior students (e.g., high school and undergraduate) to validate basic yet fundamental hypotheses considered to be uninteresting to domain experts. Here, we, a group of high school-aged students and their mentors, present the results of our participation in Grand Challenge 4 where we predicted ligand affinity rankings for the Cathepsin S protease, an important protein target for autoimmune diseases. To investigate the effect of incorporating receptor dynamics on ligand affinity rankings, we employed the Relaxed Complex Scheme, a molecular docking method paired with molecular dynamics-generated receptor conformations. We found that Cathepsin S is a difficult target for molecular docking and we explore some advanced methods such as distance-restrained docking to try to improve the correlation with experiments. This project has exemplified the capabilities of high school students when supported with a rigorous curriculum, and demonstrates the value of community-driven competitions for beginners in computational drug discovery.
新药的发现是一个耗时且昂贵的过程。虚拟筛选等方法,即在昂贵的实验研究之前,可以从药物库中筛选出无效的化合物,已成为热门的研究课题。随着计算药物发现领域的发展,为了对各种方法的进展进行基准测试,像药物设计数据资源这样的组织已经开始举办盲目的大型挑战赛,旨在确定配体构象预测、配体亲和力排序和自由能计算的最佳方法。这种公开的挑战为研究人员提供了一个独特的机会,可以与初级学生(例如高中生和本科生)合作,验证被认为对领域专家无趣的基本但基本的假设。在这里,我们是一群高中生及其导师,展示了我们参与 Cathepsin S 蛋白酶配体亲和力排序预测的结果,Cathepsin S 是一种重要的自身免疫性疾病蛋白质靶标。为了研究纳入受体动力学对配体亲和力排序的影响,我们采用了 Relaxed Complex Scheme,这是一种与基于分子动力学生成的受体构象配对的分子对接方法。我们发现 Cathepsin S 是分子对接的一个难题,我们探索了一些高级方法,例如距离约束对接,以尝试提高与实验的相关性。这个项目展示了在严格的课程支持下,高中生的能力,并证明了社区驱动的竞赛对于计算药物发现初学者的价值。