Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, Pharmacometrics & System Pharmacology PharmacoAnalytics, School of Pharmacy, University of Pittsburgh, Pittsburgh, PA 15261, USA.
NIH National Center of Excellence for Computational Drug Abuse Research (CDAR), University of Pittsburgh, Pittsburgh, PA 15261, USA.
Cells. 2022 Mar 7;11(5):915. doi: 10.3390/cells11050915.
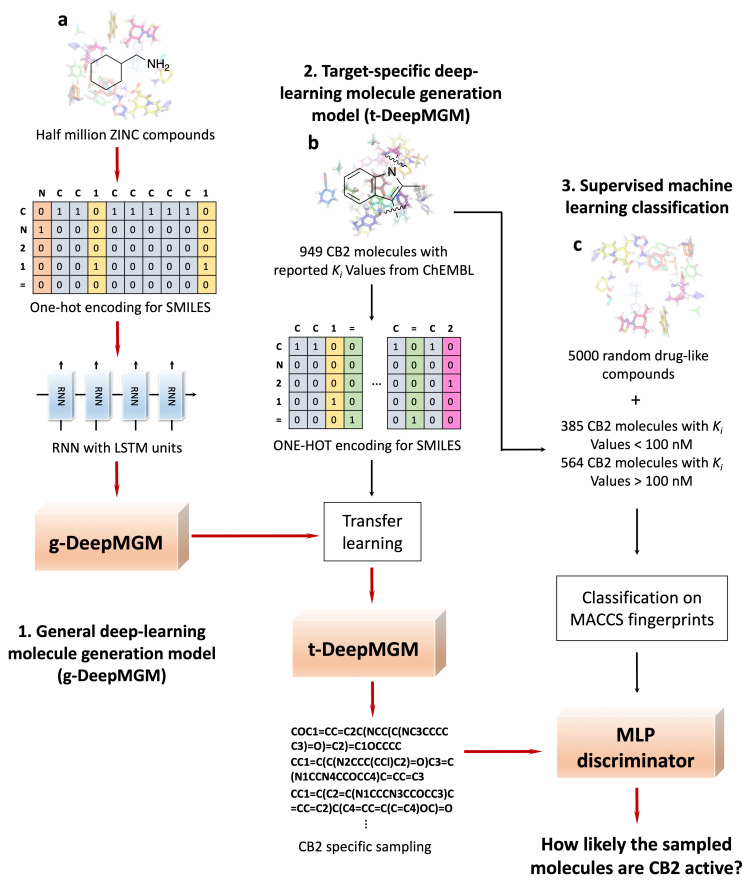
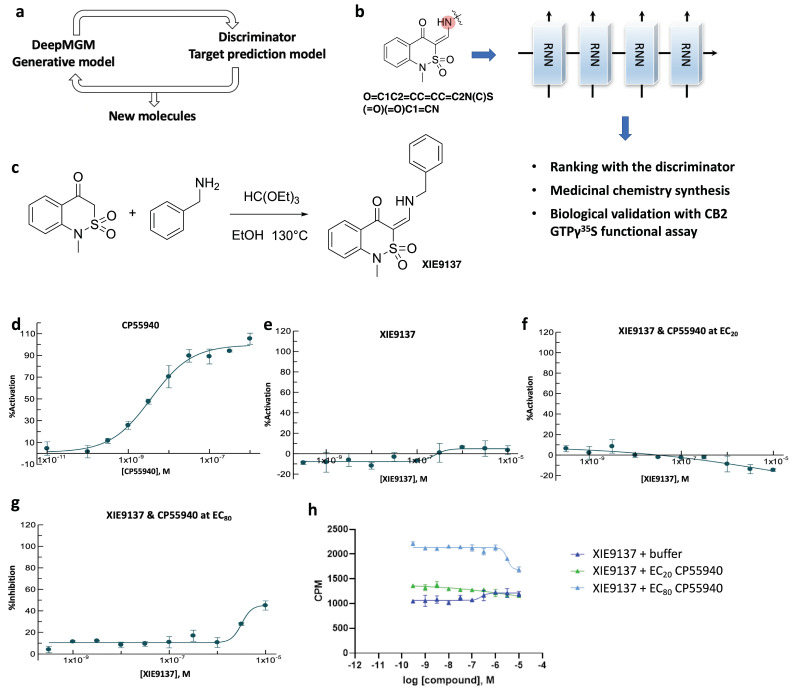
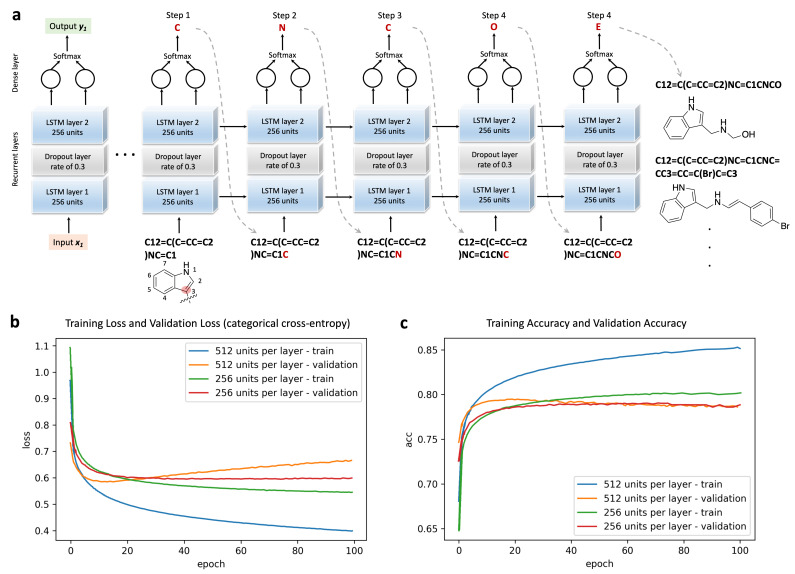
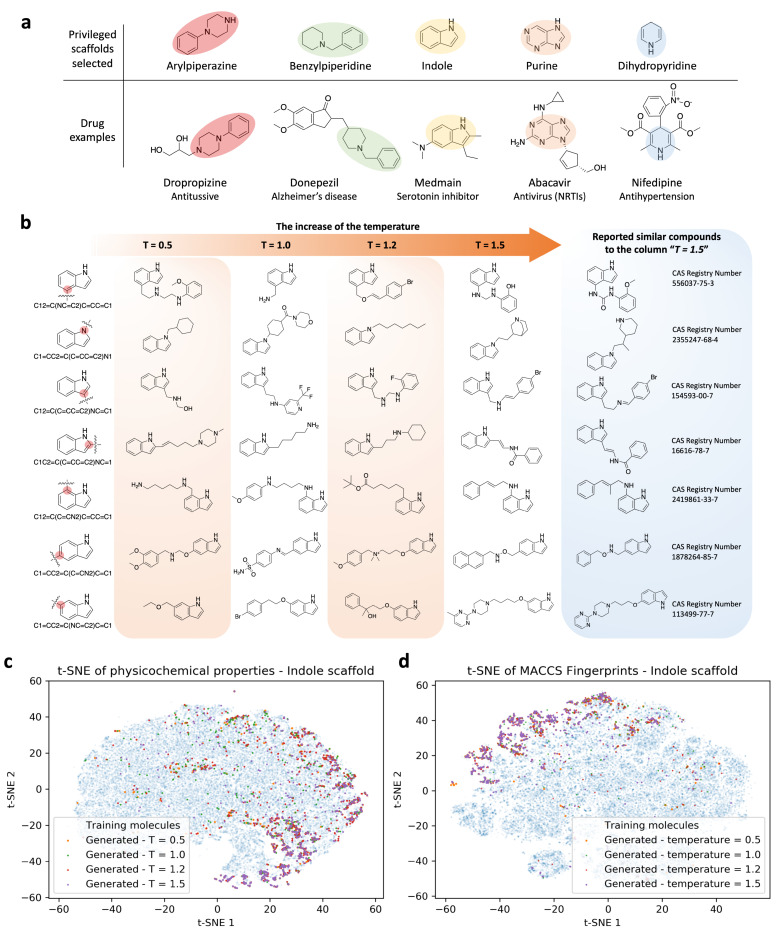
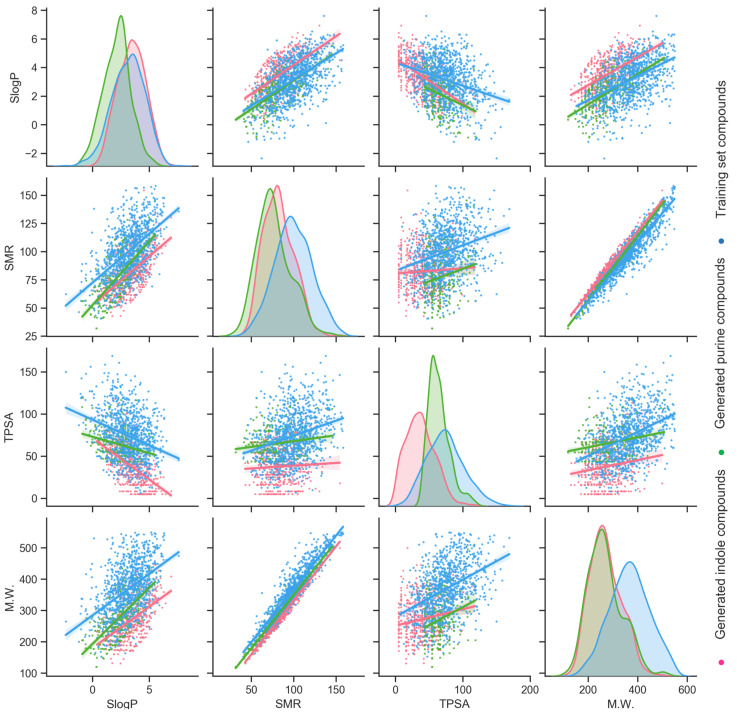
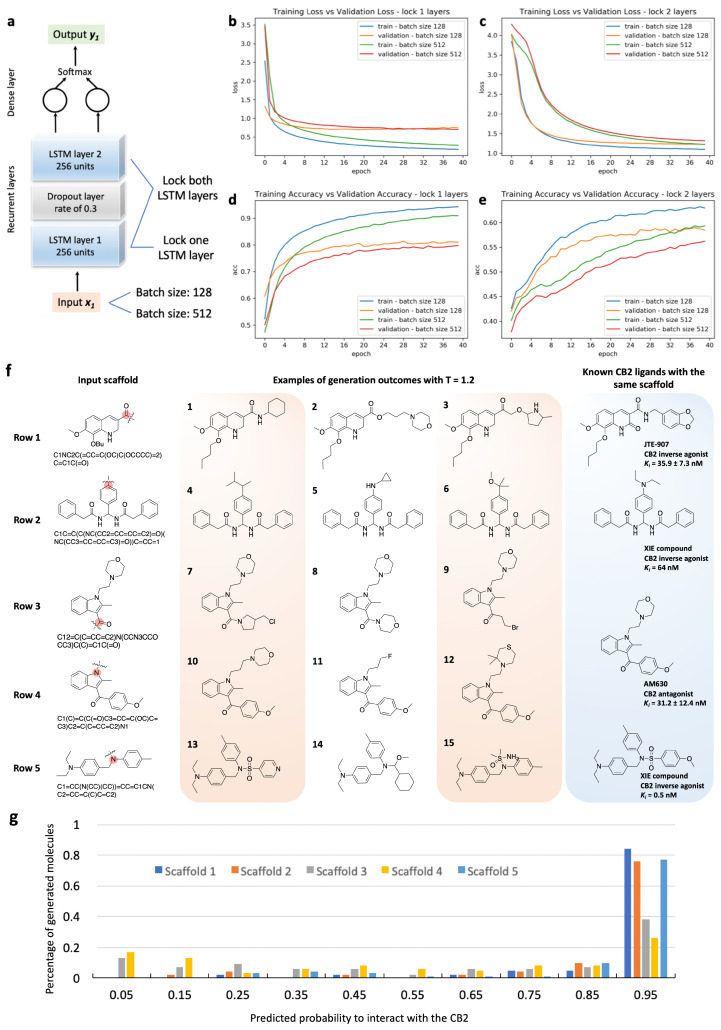
Design and generation of high-quality target- and scaffold-specific small molecules is an important strategy for the discovery of unique and potent bioactive drug molecules. To achieve this goal, authors have developed the deep-learning molecule generation model (DeepMGM) and applied it for the de novo molecular generation of scaffold-focused small-molecule libraries. In this study, a recurrent neural network (RNN) using long short-term memory (LSTM) units was trained with drug-like molecules to result in a general model (g-DeepMGM). Sampling practices on indole and purine scaffolds illustrate the feasibility of creating scaffold-focused chemical libraries based on machine intelligence. Subsequently, a target-specific model (t-DeepMGM) for cannabinoid receptor 2 (CB2) was constructed following the transfer learning process of known CB2 ligands. Sampling outcomes can present similar properties to the reported active molecules. Finally, a discriminator was trained and attached to the DeepMGM to result in an in silico molecular design-test circle. Medicinal chemistry synthesis and biological validation was performed to further investigate the generation outcome, showing that XIE9137 was identified as a potential allosteric modulator of CB2. This study demonstrates how recent progress in deep learning intelligence can benefit drug discovery, especially in de novo molecular design and chemical library generation.
设计和生成高质量的靶标和支架特异性小分子是发现独特而有效的生物活性药物分子的重要策略。为了实现这一目标,作者开发了深度学习分子生成模型(DeepMGM),并将其应用于支架聚焦小分子库的从头分子生成。在这项研究中,使用长短期记忆(LSTM)单元的递归神经网络(RNN)对类似药物的分子进行了训练,从而得到了一个通用模型(g-DeepMGM)。对吲哚和嘌呤支架的采样实践说明了基于机器智能创建支架聚焦化学库的可行性。随后,通过已知 CB2 配体的迁移学习过程,构建了针对大麻素受体 2(CB2)的靶向特异性模型(t-DeepMGM)。采样结果可以呈现出与报道的活性分子相似的性质。最后,训练了一个鉴别器并将其附加到 DeepMGM 上,以实现一个虚拟分子设计-测试循环。进行了药物化学合成和生物验证,以进一步研究生成结果,结果表明 XIE9137 被鉴定为 CB2 的潜在别构调节剂。本研究展示了深度学习智能的最新进展如何有益于药物发现,特别是在从头分子设计和化学库生成方面。