icddr, b, Formerly International Centre for Diarrhoeal Disease Research, Bangladesh, Dhaka, Bangladesh.
National Institutes of Infectious Diseases (NIID), Tokyo, Japan.
Microbiol Spectr. 2022 Apr 27;10(2):e0039122. doi: 10.1128/spectrum.00391-22. Epub 2022 Mar 22.
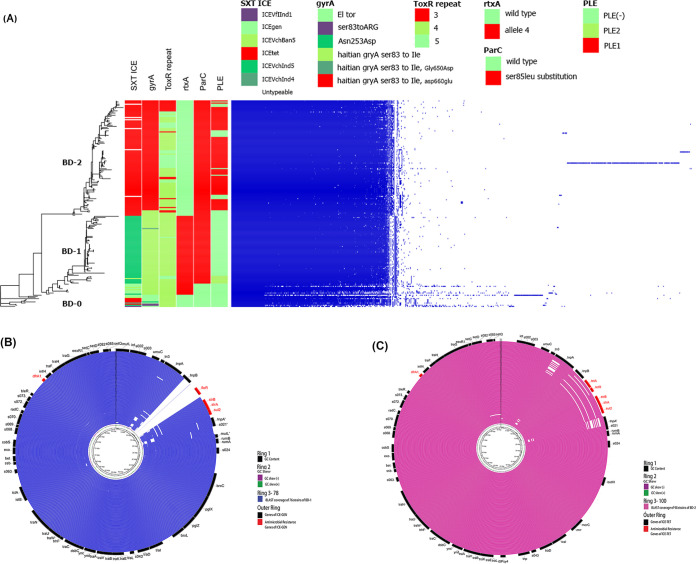
Comparative genomic analysis of Vibrio cholerae El Tor associated with endemic cholera in Asia revealed two distinct lineages, one dominant in Bangladesh and the other in India. An in-depth whole-genome study of V. cholerae El Tor strains isolated during endemic cholera in Bangladesh (1991 to 2017) included reference genome sequence data obtained online. Core genome phylogeny established using single nucleotide polymorphisms (SNPs) showed V. cholerae El Tor strains comprised two lineages, BD-1 and BD-2, which, according to Bayesian phylodynamic analysis, originated from paraphyletic group BD-0 around 1981. BD-1 and BD-2 lineages overlapped temporally but were negatively associated as causative agents of cholera during 2004 to 2017. Genome-wide association study (GWAS) revealed 140 SNPs and 31 indels, resulting in gene alleles unique to BD-1 and BD-2. Regression analysis of root to tip distance and year of isolation indicated early BD-0 strains at the base, whereas BD-1 and BD-2 subsequently emerged and progressed by accumulating SNPs. Pangenome analysis provided evidence of gene acquisition by both BD-1 and BD-2, of which six crucial proteins of known function were predominant in BD-2. BD-1 and BD-2 diverged and have distinctively different genomic traits, namely, heterogeneity in VSP-2, VPI-1, mobile elements, toxin encoding elements, and total gene abundance. In addition, the observed phage-inducible chromosomal island-like element (PLE1), and SXT ICE elements (ICE) in BD-2 presumably provided a fitness advantage for the lineage to outcompete BD-1 as the etiological agent of endemic cholera in Bangladesh, with implications for global cholera epidemiology. Cholera is a global disease with specific reference to the Bay of Bengal Ganges Delta where Vibrio cholerae O1 El Tor, the causative agent of the disease showed two circulating lineages, one dominant in Bangladesh and the other in India. Results of an in-depth genomic study of V. cholerae associated with endemic cholera during the past 27 years (1991 to 2017) indicate emergence and succession of the two lineages, BD-1 and BD-2, arising from a common ancestral paraphyletic group, BD-0, comprising the early strains and short-term evolution of the bacterium in Bangladesh. Among the two V. cholerae lineages, BD-2 supersedes BD-1 and is predominant in the most recent endemic cholera in Bangladesh. The BD-2 lineage contained significantly more SNPs and indels, and showed richness in gene abundance, including antimicrobial resistance genes, gene cassettes, and PLE to fight against bacteriophage infection, acquired over time. These findings have important epidemic implications on a global scale.
比较分析与亚洲地方性霍乱相关的霍乱弧菌 El Tor ,揭示了两个不同的谱系,一个在孟加拉国占主导地位,另一个在印度。对孟加拉国地方性霍乱期间(1991 年至 2017 年)分离的霍乱弧菌 El Tor 菌株进行了深入的全基因组研究,包括在线获得的参考基因组序列数据。使用单核苷酸多态性(SNP)建立的核心基因组系统发育表明,霍乱弧菌 El Tor 菌株由两个谱系组成,BD-1 和 BD-2,根据贝叶斯系统发育分析,它们起源于 1981 年左右的并系群 BD-0。BD-1 和 BD-2 谱系在时间上重叠,但在 2004 年至 2017 年期间作为霍乱的病原体呈负相关。全基因组关联研究(GWAS)显示了 140 个 SNP 和 31 个插入缺失,导致 BD-1 和 BD-2 特有的基因等位基因。根到尖距离和分离年份的回归分析表明,早期的 BD-0 菌株位于底部,而随后 BD-1 和 BD-2 出现并通过积累 SNP 而进化。泛基因组分析提供了 BD-1 和 BD-2 获得基因的证据,其中已知功能的六个关键蛋白在 BD-2 中占主导地位。BD-1 和 BD-2 分化并具有明显不同的基因组特征,即 VSP-2、VPI-1、移动元件、毒素编码元件和总基因丰度的异质性。此外,观察到的噬菌体诱导的染色体岛样元件(PLE1)和 SXT ICE 元件(ICE)在 BD-2 中,可能为该谱系提供了适应优势,使其能够在孟加拉国地方性霍乱中与 BD-1 竞争,这对全球霍乱流行病学具有重要意义。霍乱是一种全球性疾病,特别是在孟加拉湾恒河三角洲,那里的霍乱弧菌 O1 El Tor 是该病的病原体,表现出两种循环谱系,一种在孟加拉国占主导地位,另一种在印度。过去 27 年(1991 年至 2017 年)与地方性霍乱相关的霍乱弧菌的深入基因组研究结果表明,BD-1 和 BD-2 这两个谱系的出现和接替,源于一个共同的祖先并系群 BD-0,包括早期菌株和孟加拉国细菌的短期进化。在两个霍乱弧菌谱系中,BD-2 取代了 BD-1,在孟加拉国最近的地方性霍乱中占主导地位。BD-2 谱系包含了更多的 SNP 和插入缺失,显示了基因丰度的丰富性,包括抗菌药物抗性基因、基因盒和 PLE 以抵抗噬菌体感染,这些都是随着时间的推移获得的。这些发现对全球范围内的疫情具有重要的影响。