Department of Biochemistry, University of Washington, Seattle, WA, USA.
Institute for Protein Design, University of Washington, Seattle, WA, USA.
Nature. 2022 May;605(7910):551-560. doi: 10.1038/s41586-022-04654-9. Epub 2022 Mar 24.
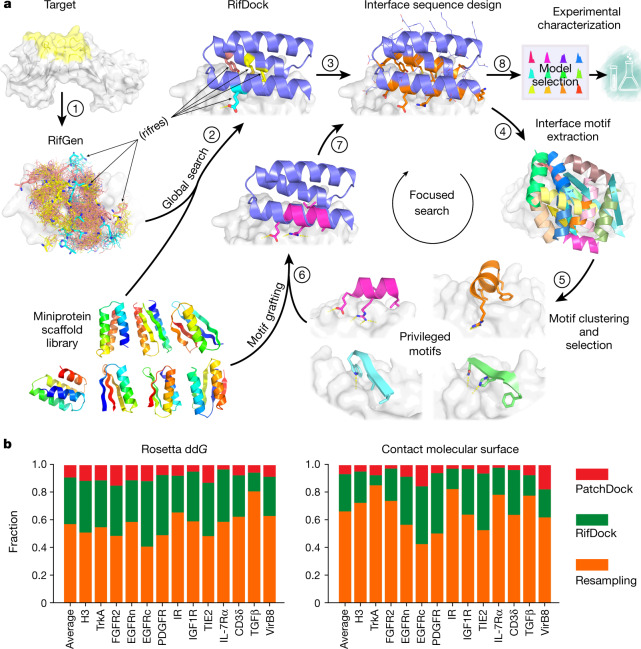
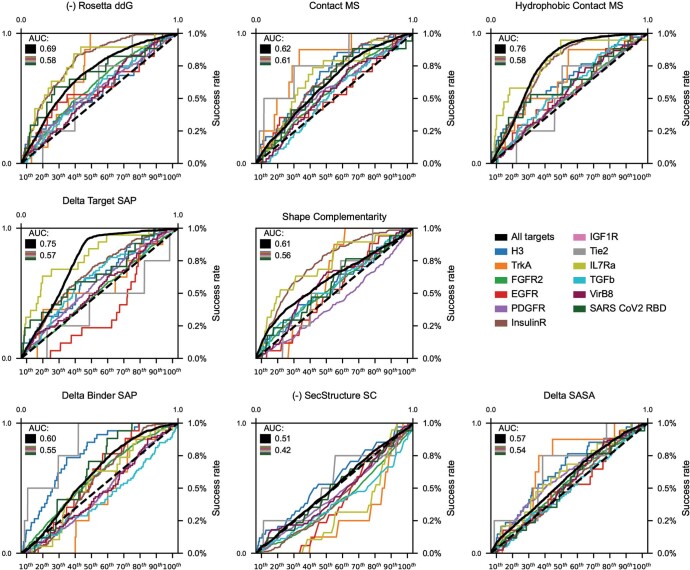
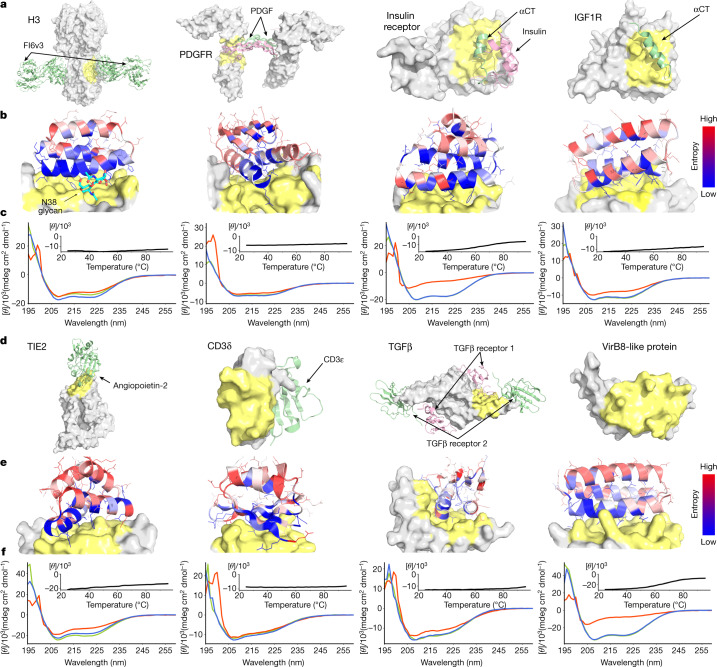
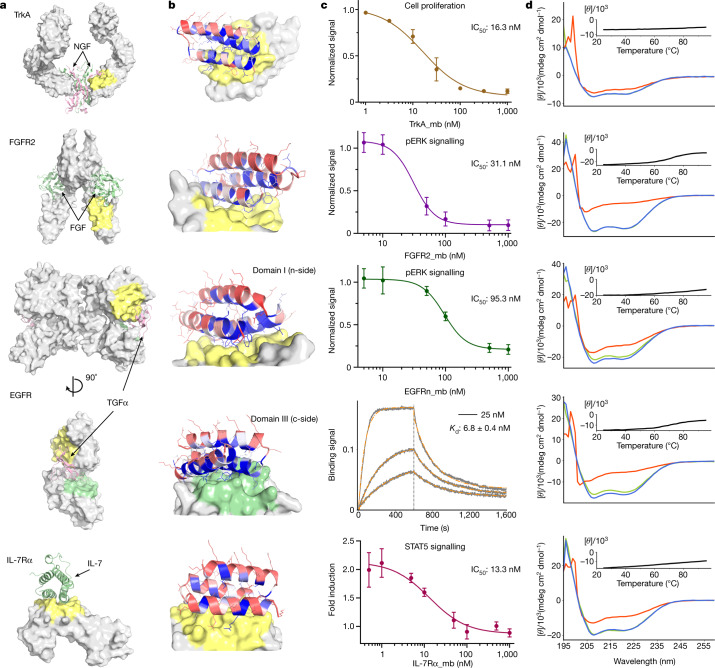
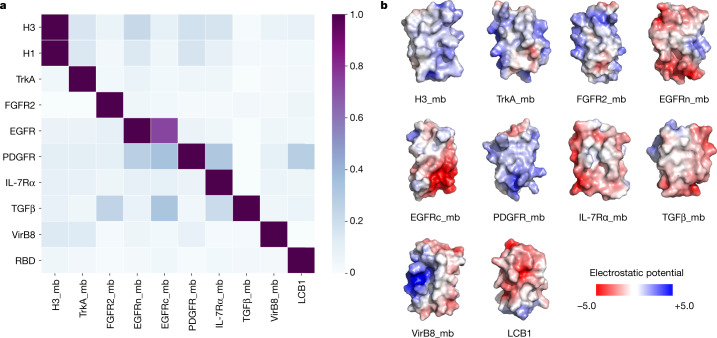
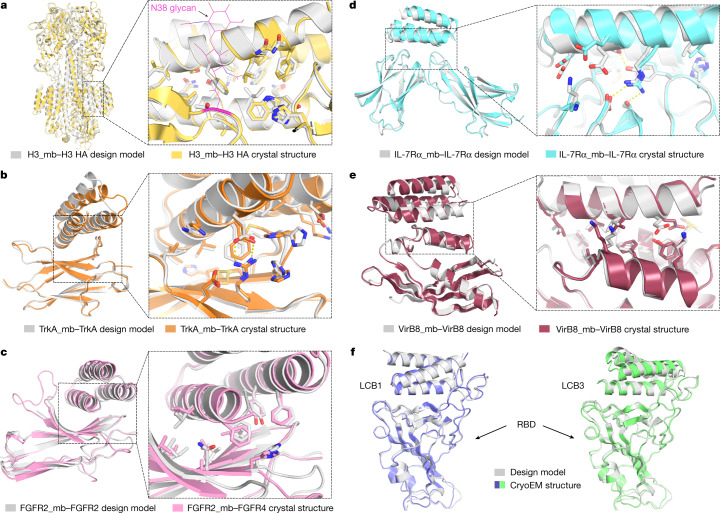
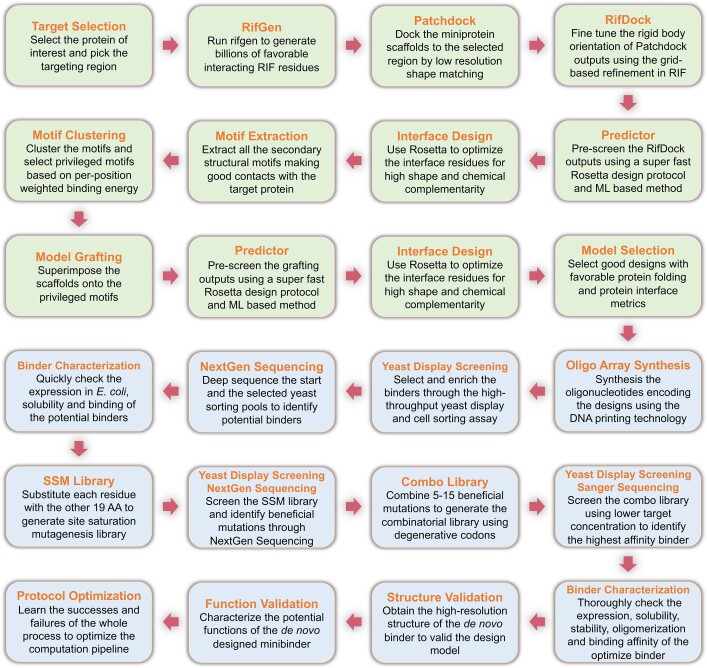
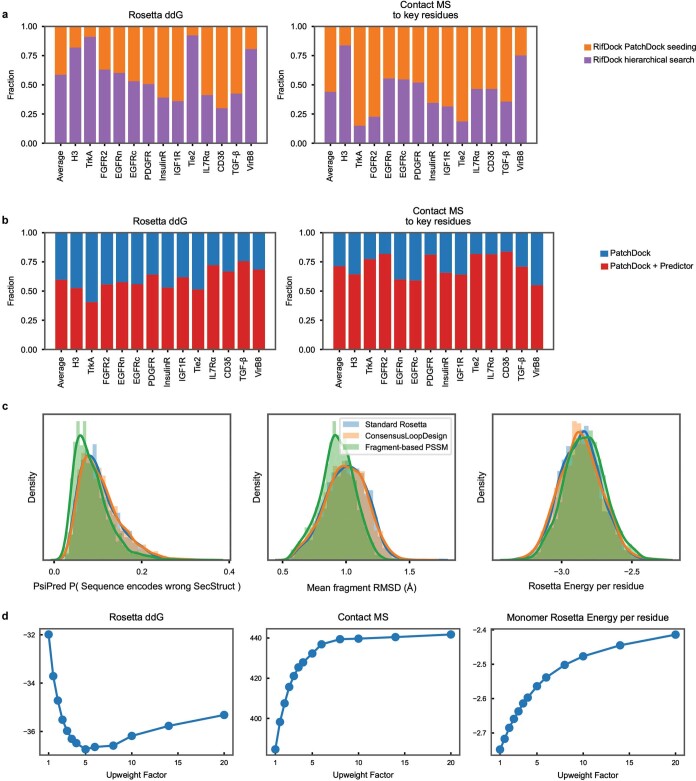
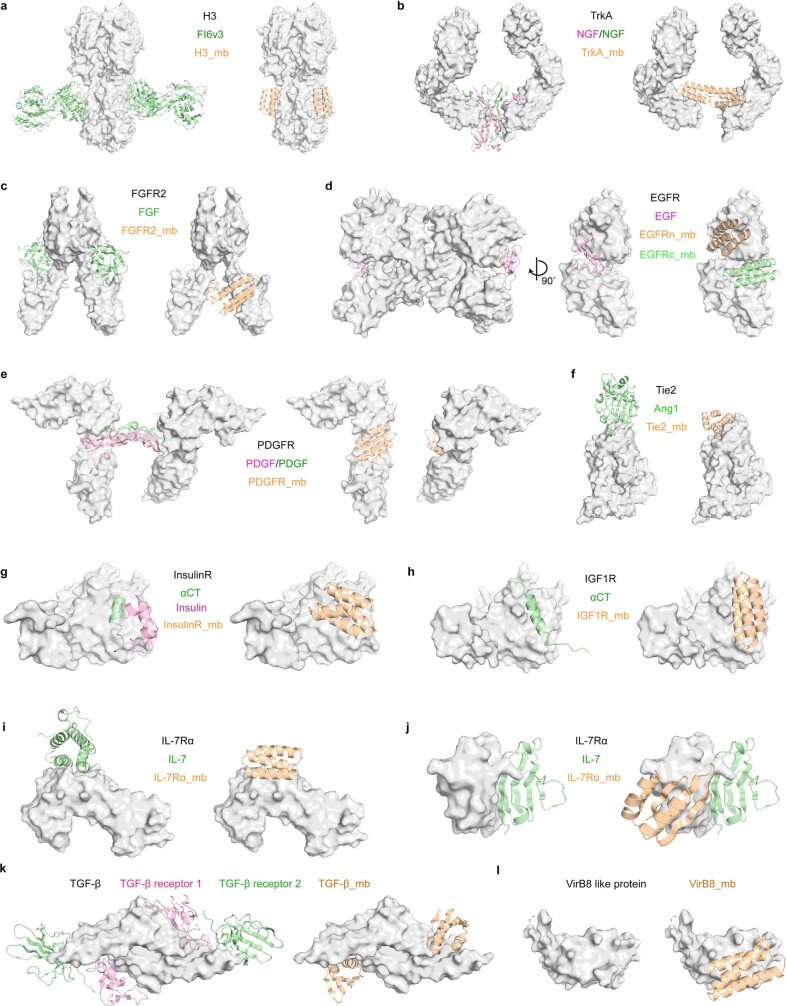
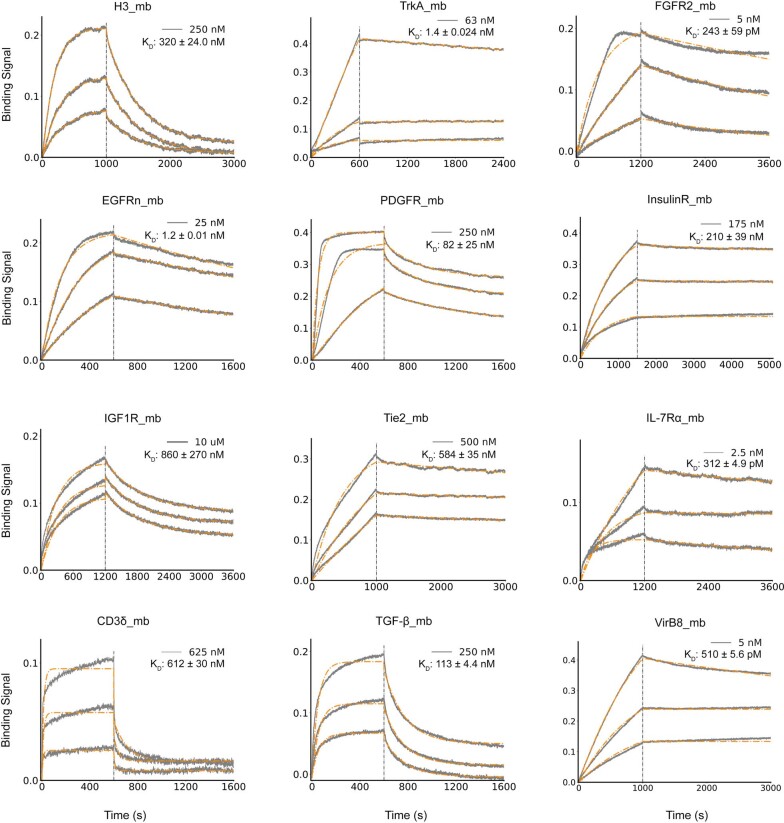
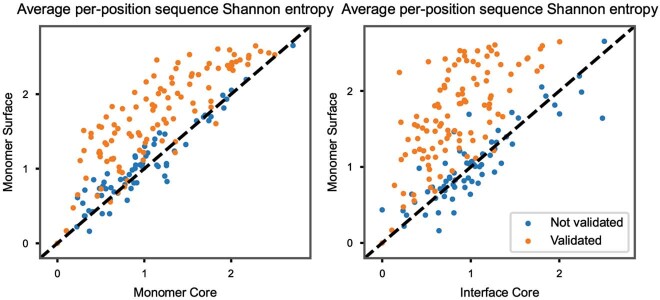
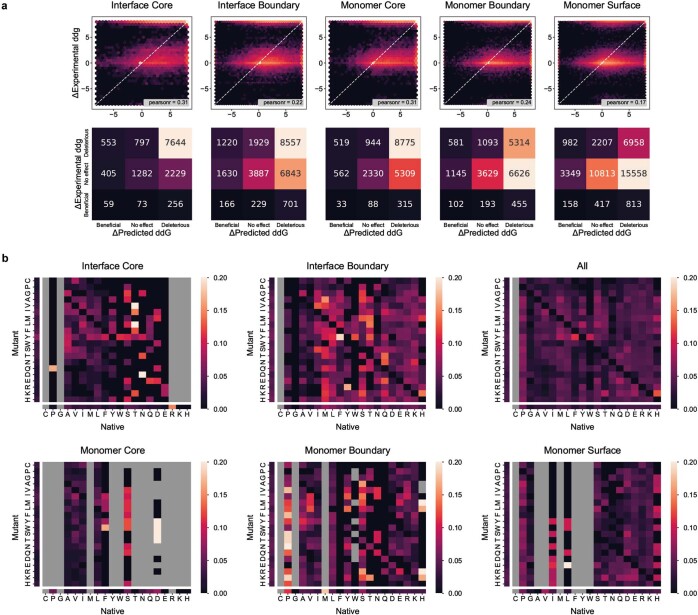
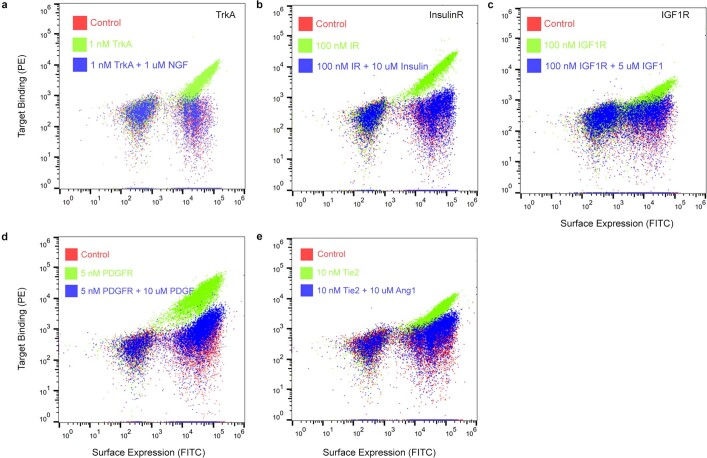
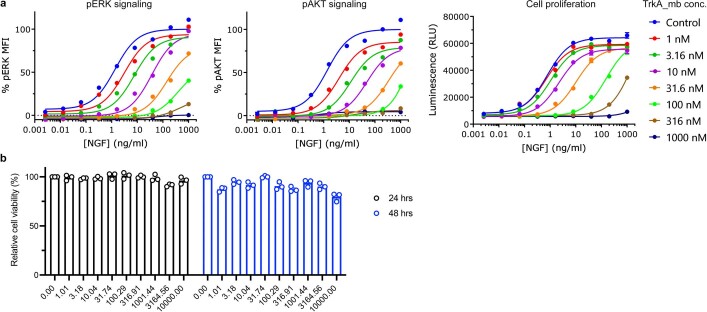
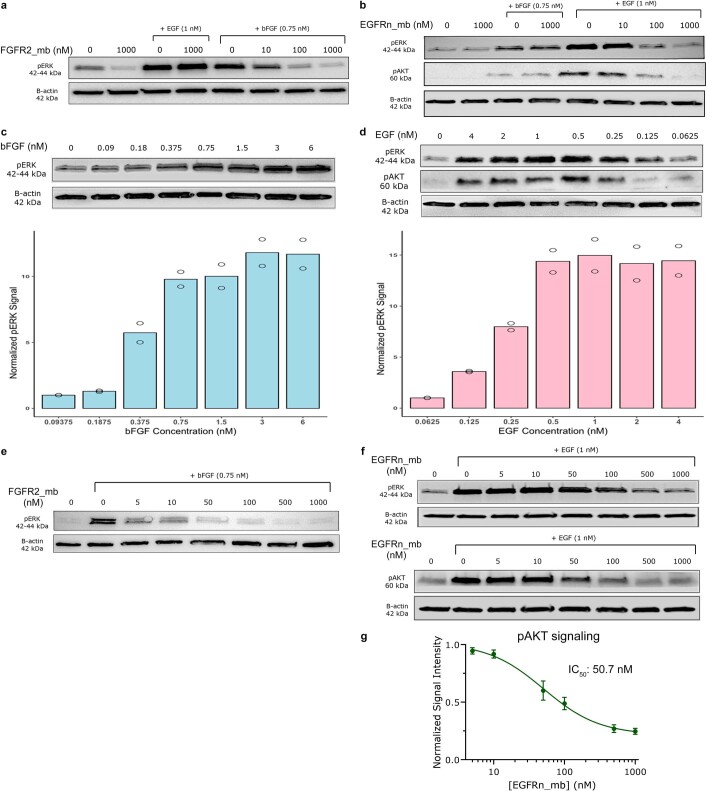
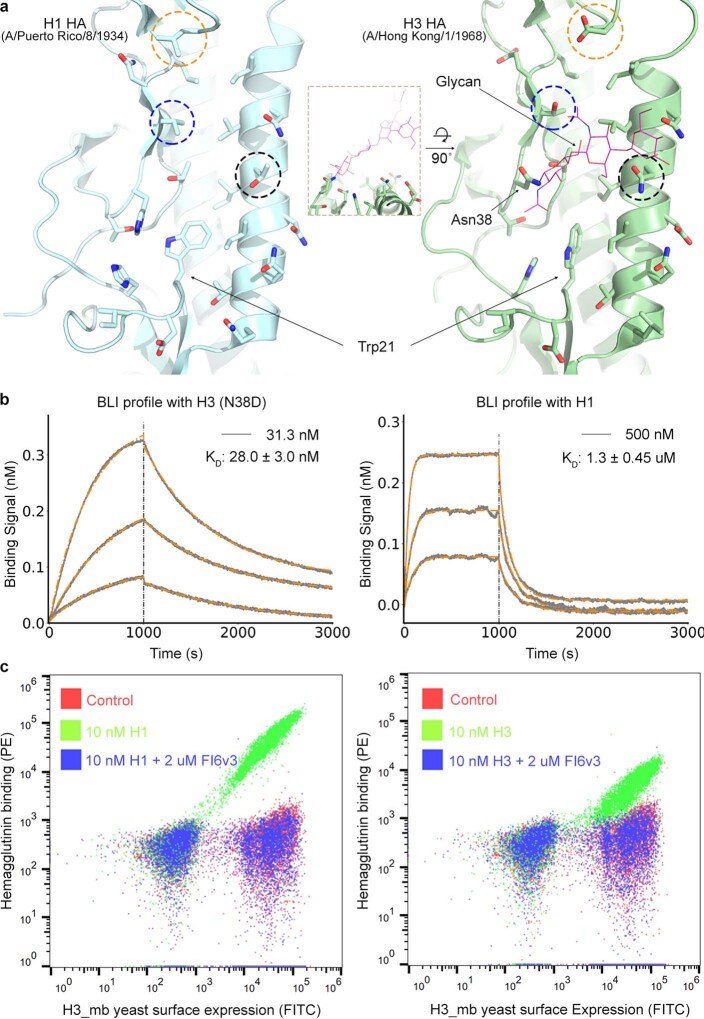
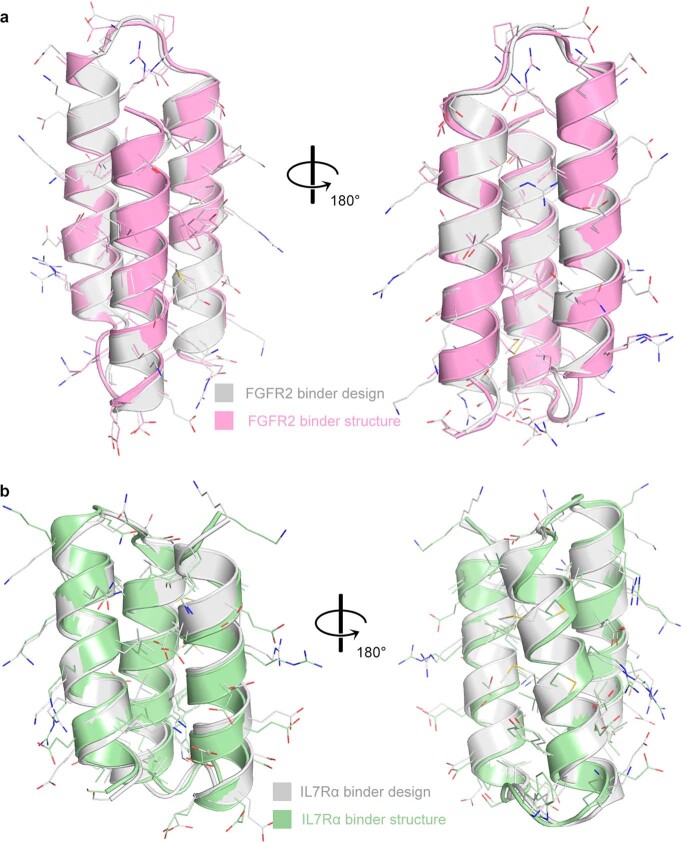
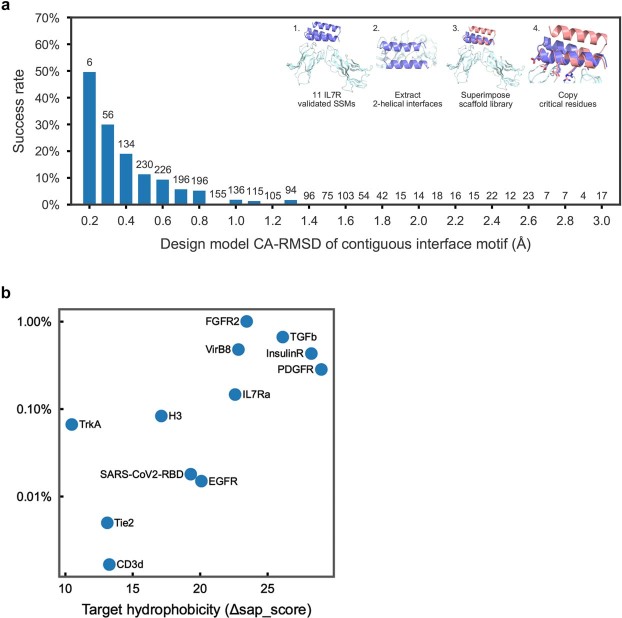
The design of proteins that bind to a specific site on the surface of a target protein using no information other than the three-dimensional structure of the target remains a challenge. Here we describe a general solution to this problem that starts with a broad exploration of the vast space of possible binding modes to a selected region of a protein surface, and then intensifies the search in the vicinity of the most promising binding modes. We demonstrate the broad applicability of this approach through the de novo design of binding proteins to 12 diverse protein targets with different shapes and surface properties. Biophysical characterization shows that the binders, which are all smaller than 65 amino acids, are hyperstable and, following experimental optimization, bind their targets with nanomolar to picomolar affinities. We succeeded in solving crystal structures of five of the binder-target complexes, and all five closely match the corresponding computational design models. Experimental data on nearly half a million computational designs and hundreds of thousands of point mutants provide detailed feedback on the strengths and limitations of the method and of our current understanding of protein-protein interactions, and should guide improvements of both. Our approach enables the targeted design of binders to sites of interest on a wide variety of proteins for therapeutic and diagnostic applications.
使用除目标蛋白质表面的三维结构之外的任何信息来设计与目标蛋白质表面的特定部位结合的蛋白质仍然是一个挑战。在这里,我们描述了一个通用的解决方案,该方案从对蛋白质表面选定区域的各种可能结合模式的广泛探索开始,然后在最有希望的结合模式附近加强搜索。我们通过从头设计与 12 种具有不同形状和表面特性的蛋白质靶标的结合蛋白来证明该方法的广泛适用性。生物物理特性表明,这些结合物均小于 65 个氨基酸,其稳定性很高,经过实验优化后,其对靶标的亲和力达到纳摩尔到皮摩尔级。我们成功地解决了其中五个结合物-靶标复合物的晶体结构,并且这五个结构都与相应的计算设计模型非常吻合。将近 50 万个计算设计和数十万个点突变的实验数据为该方法的优缺点以及我们目前对蛋白质-蛋白质相互作用的理解提供了详细的反馈,并且应该指导两者的改进。我们的方法能够针对各种蛋白质上的感兴趣的部位进行靶向设计结合物,用于治疗和诊断应用。