He Jiazhen, Nittinger Eva, Tyrchan Christian, Czechtizky Werngard, Patronov Atanas, Bjerrum Esben Jannik, Engkvist Ola
Molecular AI, Discovery Sciences, R&D, AstraZeneca, Gothenburg, Sweden.
Medicinal Chemistry, Research and Early Development, Respiratory and Immunology (R&I), BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden.
J Cheminform. 2022 Mar 28;14(1):18. doi: 10.1186/s13321-022-00599-3.
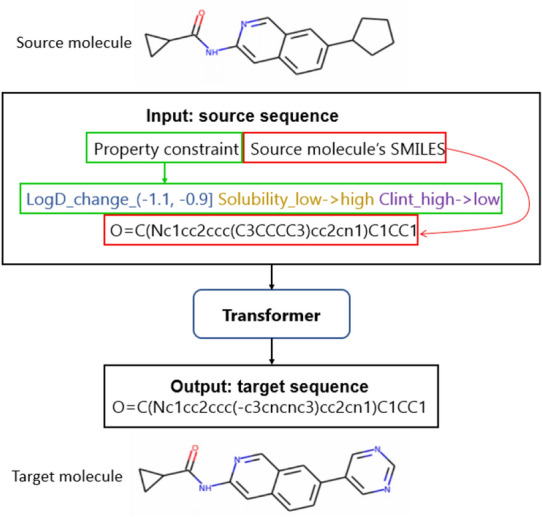
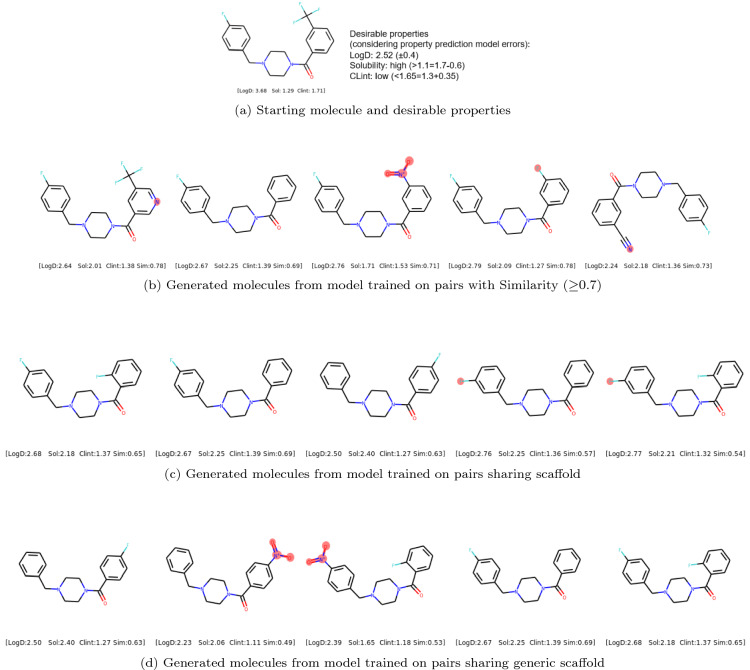
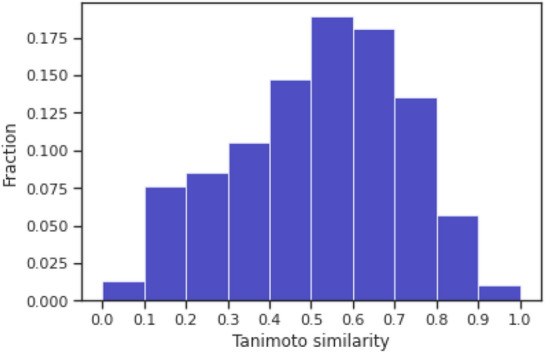
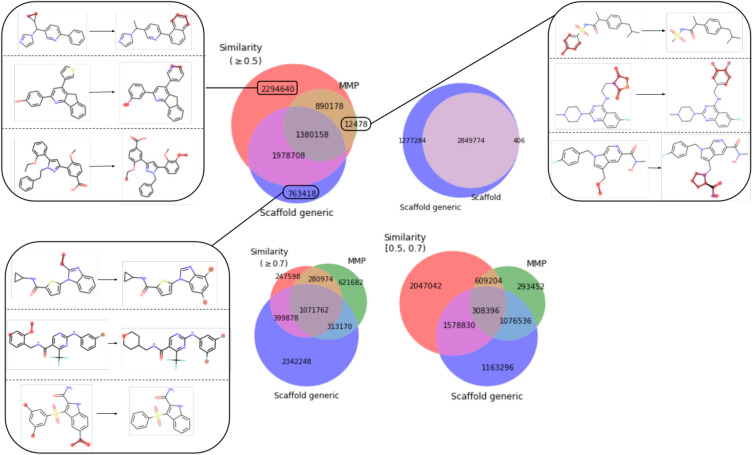
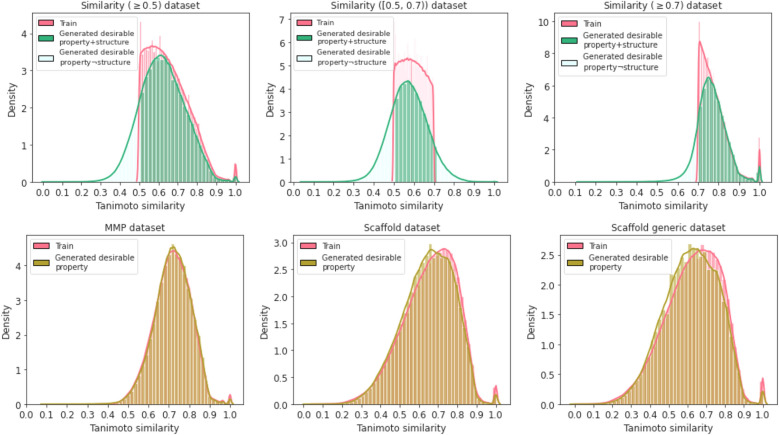
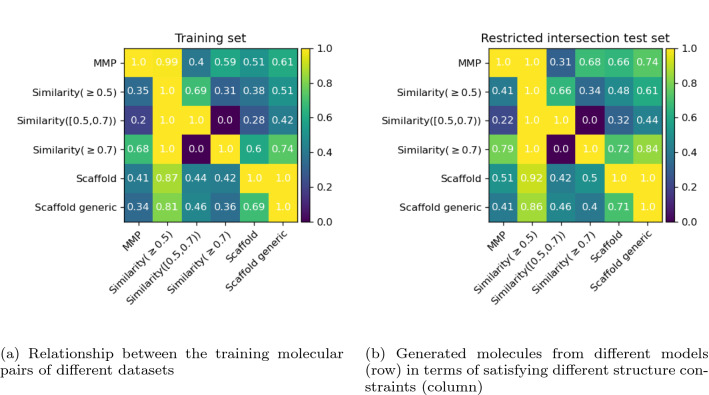
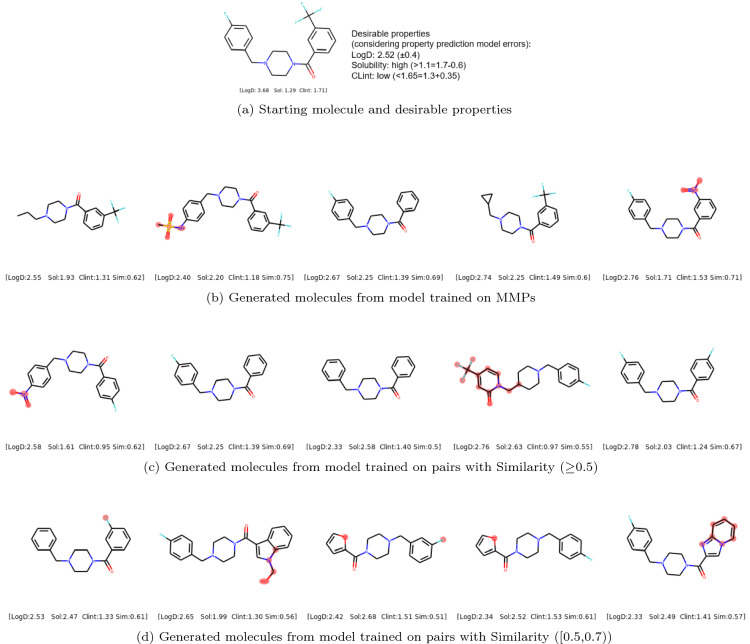
Molecular optimization aims to improve the drug profile of a starting molecule. It is a fundamental problem in drug discovery but challenging due to (i) the requirement of simultaneous optimization of multiple properties and (ii) the large chemical space to explore. Recently, deep learning methods have been proposed to solve this task by mimicking the chemist's intuition in terms of matched molecular pairs (MMPs). Although MMPs is a widely used strategy by medicinal chemists, it offers limited capability in terms of exploring the space of structural modifications, therefore does not cover the complete space of solutions. Often more general transformations beyond the nature of MMPs are feasible and/or necessary, e.g. simultaneous modifications of the starting molecule at different places including the core scaffold. This study aims to provide a general methodology that offers more general structural modifications beyond MMPs. In particular, the same Transformer architecture is trained on different datasets. These datasets consist of a set of molecular pairs which reflect different types of transformations. Beyond MMP transformation, datasets reflecting general structural changes are constructed from ChEMBL based on two approaches: Tanimoto similarity (allows for multiple modifications) and scaffold matching (allows for multiple modifications but keep the scaffold constant) respectively. We investigate how the model behavior can be altered by tailoring the dataset while using the same model architecture. Our results show that the models trained on differently prepared datasets transform a given starting molecule in a way that it reflects the nature of the dataset used for training the model. These models could complement each other and unlock the capability for the chemists to pursue different options for improving a starting molecule.
分子优化旨在改善起始分子的药物特性。这是药物发现中的一个基本问题,但具有挑战性,原因如下:(i)需要同时优化多种特性;(ii)需要探索的化学空间很大。最近,有人提出了深度学习方法,通过模仿药物化学家在匹配分子对(MMP)方面的直觉来解决这项任务。尽管MMP是药物化学家广泛使用的一种策略,但在探索结构修饰空间方面能力有限,因此并未涵盖完整的解决方案空间。通常,超出MMP性质的更一般的转化是可行的和/或必要的,例如在不同位置(包括核心支架)同时对起始分子进行修饰。本研究旨在提供一种通用方法,该方法能提供超出MMP范围的更一般的结构修饰。具体而言,在不同数据集上训练相同的Transformer架构。这些数据集由一组反映不同类型转化的分子对组成。除了MMP转化之外,基于两种方法从ChEMBL构建反映一般结构变化的数据集:分别是Tanimoto相似性(允许多重修饰)和支架匹配(允许多重修饰但保持支架不变)。我们研究了在使用相同模型架构的情况下,如何通过定制数据集来改变模型行为。我们的结果表明,在不同制备的数据集上训练的模型以一种反映用于训练模型的数据集性质的方式转化给定的起始分子。这些模型可以相互补充,为化学家提供更多选择,以改进起始分子。