School of Life Sciences, Arizona State Universitygrid.215654.1, Tempe, Arizona, USA.
Center for Fundamental and Applied Microbiomics, Arizona State Universitygrid.215654.1, Tempe, Arizona, USA.
mSystems. 2022 Apr 26;7(2):e0016722. doi: 10.1128/msystems.00167-22. Epub 2022 Apr 4.
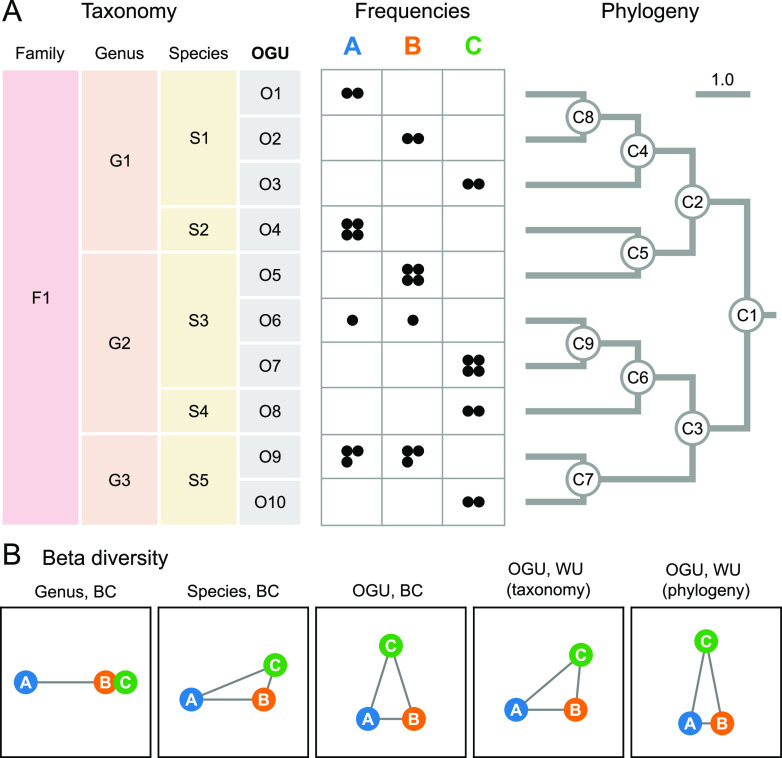
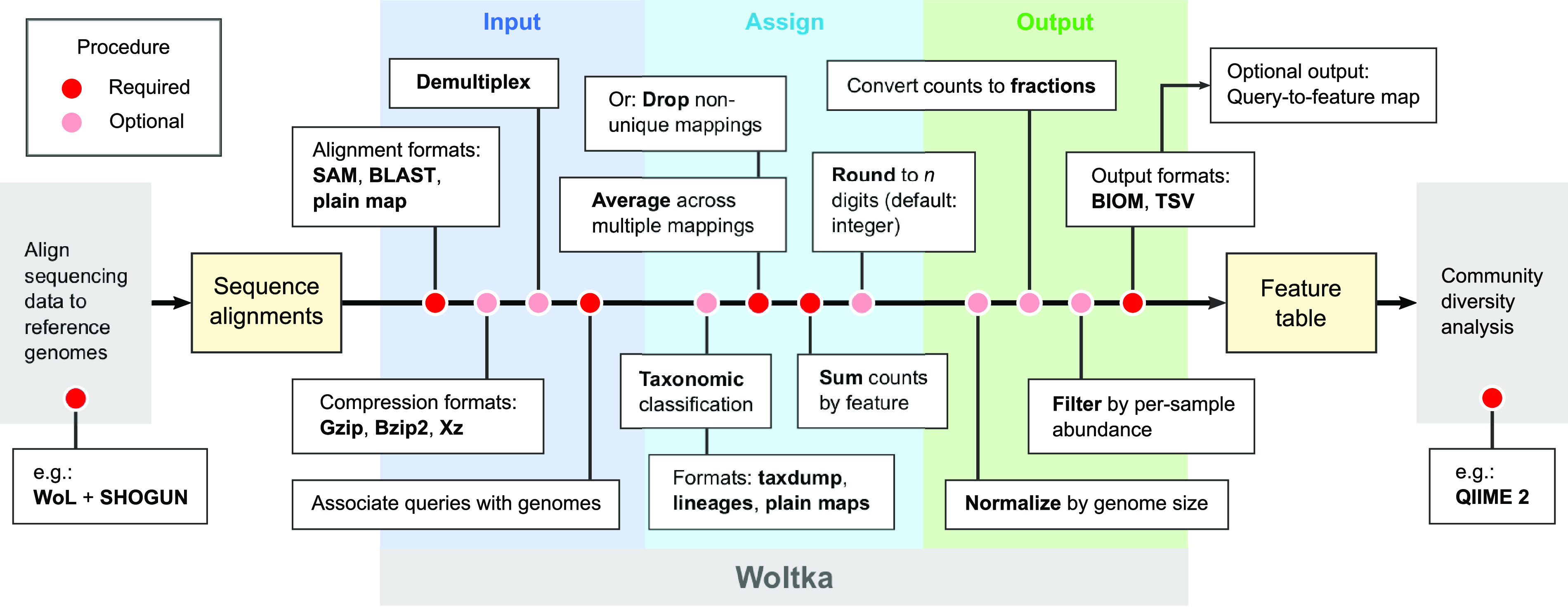
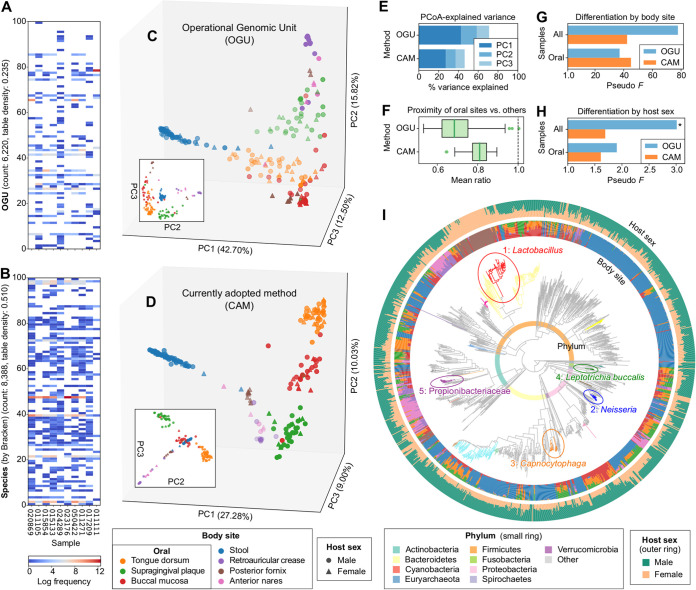
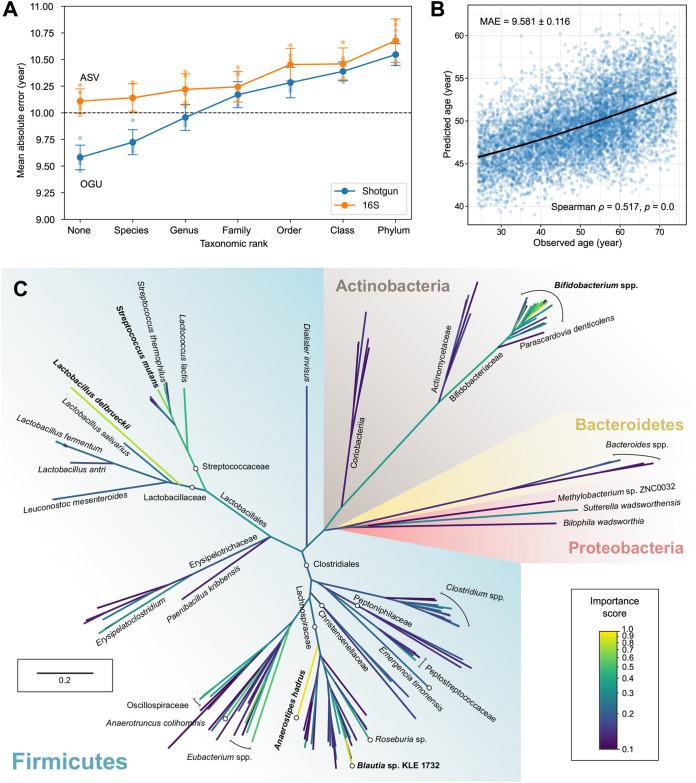
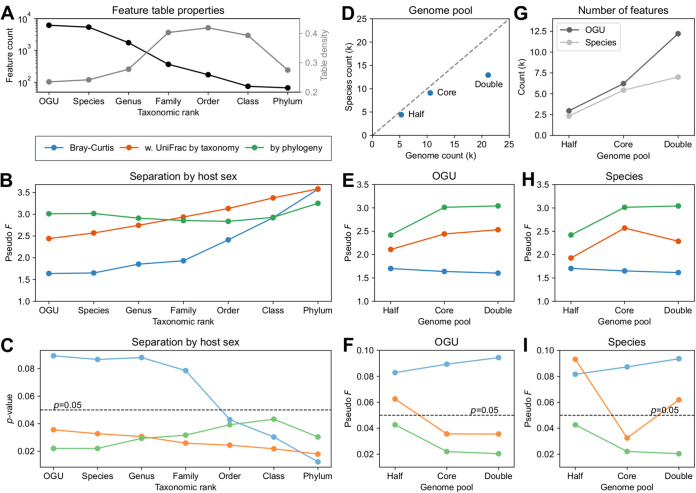
We introduce the operational genomic unit (OGU) method, a metagenome analysis strategy that directly exploits sequence alignment hits to individual reference genomes as the minimum unit for assessing the diversity of microbial communities and their relevance to environmental factors. This approach is independent of taxonomic classification, granting the possibility of maximal resolution of community composition, and organizes features into an accurate hierarchy using a phylogenomic tree. The outputs are suitable for contemporary analytical protocols for community ecology, differential abundance, and supervised learning while supporting phylogenetic methods, such as UniFrac and phylofactorization, that are seldom applied to shotgun metagenomics despite being prevalent in 16S rRNA gene amplicon studies. As demonstrated in two real-world case studies, the OGU method produces biologically meaningful patterns from microbiome data sets. Such patterns further remain detectable at very low metagenomic sequencing depths. Compared with taxonomic unit-based analyses implemented in currently adopted metagenomics tools, and the analysis of 16S rRNA gene amplicon sequence variants, this method shows superiority in informing biologically relevant insights, including stronger correlation with body environment and host sex on the Human Microbiome Project data set and more accurate prediction of human age by the gut microbiomes of Finnish individuals included in the FINRISK 2002 cohort. We provide Woltka, a bioinformatics tool to implement this method, with full integration with the QIIME 2 package and the Qiita web platform, to facilitate adoption of the OGU method in future metagenomics studies. Shotgun metagenomics is a powerful, yet computationally challenging, technique compared to 16S rRNA gene amplicon sequencing for decoding the composition and structure of microbial communities. Current analyses of metagenomic data are primarily based on taxonomic classification, which is limited in feature resolution. To solve these challenges, we introduce operational genomic units (OGUs), which are the individual reference genomes derived from sequence alignment results, without further assigning them taxonomy. The OGU method advances current read-based metagenomics in two dimensions: (i) providing maximal resolution of community composition and (ii) permitting use of phylogeny-aware tools. Our analysis of real-world data sets shows that it is advantageous over currently adopted metagenomic analysis methods and the finest-grained 16S rRNA analysis methods in predicting biological traits. We thus propose the adoption of OGUs as an effective practice in metagenomic studies.
我们介绍了操作基因组单元(OGU)方法,这是一种宏基因组分析策略,它直接利用序列比对命中单个参考基因组作为评估微生物群落多样性及其与环境因素相关性的最小单位。这种方法独立于分类学分类,具有最大程度解析群落组成的可能性,并使用系统发育树将特征组织成准确的层次结构。输出结果适用于当前的群落生态学分析协议、差异丰度和有监督学习,同时支持 UniFrac 和 phylofactorization 等系统发育方法,尽管这些方法在 16S rRNA 基因扩增子研究中很普遍,但在 shotgun 宏基因组学中很少应用。正如在两个实际案例研究中所证明的那样,OGU 方法从微生物组数据集中产生了有生物学意义的模式。这种模式在非常低的宏基因组测序深度下仍然可以检测到。与当前采用的宏基因组工具中基于分类单元的分析以及 16S rRNA 基因扩增子序列变体的分析相比,该方法在提供生物学相关见解方面具有优势,包括在人类微生物组计划数据集上与身体环境和宿主性别更强的相关性,以及通过包含在 FINRISK 2002 队列中的芬兰个体的肠道微生物组更准确地预测人类年龄。我们提供了 Woltka,这是一个用于实施该方法的生物信息学工具,它与 QIIME 2 包和 Qiita 网络平台完全集成,以促进未来宏基因组学研究中对 OGU 方法的采用。与 16S rRNA 基因扩增子测序相比,shotgun 宏基因组学是一种强大但计算上具有挑战性的技术,用于解码微生物群落的组成和结构。目前的宏基因组数据分析主要基于分类学分类,其特征分辨率有限。为了解决这些挑战,我们引入了操作基因组单元(OGU),这是从序列比对结果中得出的单个参考基因组,而无需进一步对其进行分类。OGU 方法在两个方面推进了当前基于读取的宏基因组学:(i)提供最大程度的群落组成分辨率,(ii)允许使用与系统发育相关的工具。我们对真实数据集的分析表明,它优于当前采用的宏基因组分析方法和最细粒度的 16S rRNA 分析方法在预测生物特征方面的表现。因此,我们建议采用 OGU 作为宏基因组学研究中的有效实践。