Anhui Provincial Centers for Disease Control and Prevention, Hefei, China.
Department of Microbiology, The Key Laboratory of Microbiology and Parasitology of Anhui Province, The Key Laboratory of Zoonoses of High Institutions in Anhui, School of Basic Medical Sciences, Anhui Medical University, Hefei, China.
Front Cell Infect Microbiol. 2022 Mar 15;12:824578. doi: 10.3389/fcimb.2022.824578. eCollection 2022.
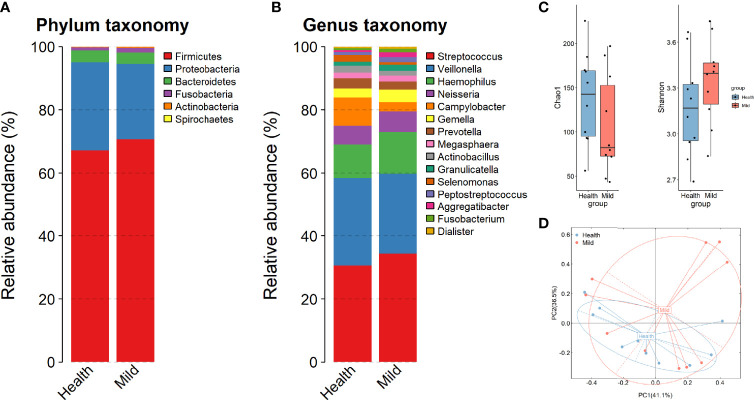
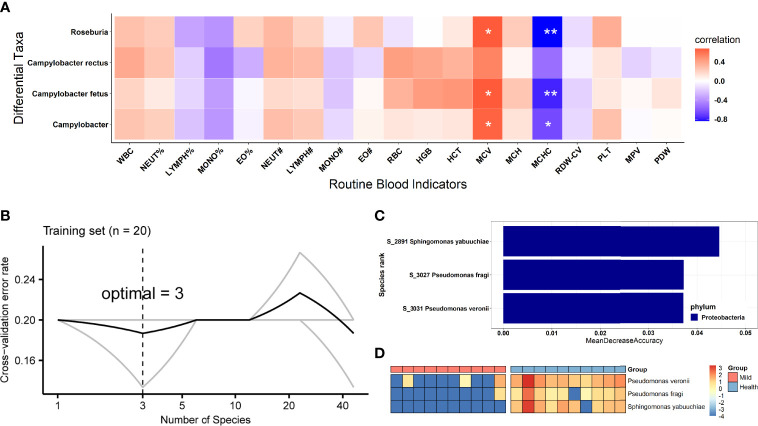
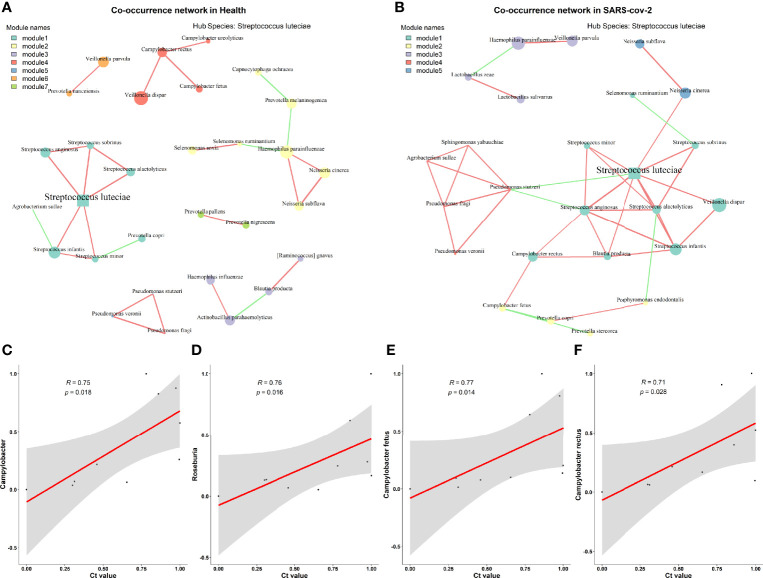
Coronavirus disease 2019 (COVID-19) remains a serious emerging global health problem, and little is known about the role of oropharynx commensal microbes in infection susceptibility and severity. Here, we present the oropharyngeal microbiota characteristics identified by full-length 16S rRNA gene sequencing through the NANOPORE platform of oropharynx swab specimens from 10 mild COVID-19 patients and 10 healthy controls. Our results revealed a distinct oropharyngeal microbiota composition in mild COVID-19 patients, characterized by enrichment of opportunistic pathogens such as and and depletion of , , and . Based on the relative abundance of the oropharyngeal microbiota at the species level, we built a microbial classifier to distinguish COVID-19 patients from healthy controls, in which , , and were identified as the most prominent signatures for their depletion in the COVID-19 group. Several members of the genus , especially and , which were highly enriched in COVID-19 patients with higher severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral load and showed a significant correlation with disease status and several routine clinical blood indicators, indicate that several bacteria may transform into opportunistic pathogen in COVID-19 patients when facing the challenges of viral infection. We also found the diver taxa and in the network of disease patients, suggesting that these oropharynx microbiota alterations may impact COVID-19 severity by influencing the microbial association patterns. In conclusion, the low sample size of SARS-CoV-2 infection patients (n = 10) here makes these results tentative; however, we have provided the overall characterization that oropharyngeal microbiota alterations and microbial correlation patterns were associated with COVID-19 severity in Anhui Province.
新型冠状病毒病(COVID-19)仍然是一个严重的全球新兴健康问题,人们对口咽共生微生物在感染易感性和严重程度中的作用知之甚少。在这里,我们通过 NANOPORE 平台对 10 例轻度 COVID-19 患者和 10 例健康对照者的口咽拭子标本进行全长 16S rRNA 基因测序,鉴定出口咽共生微生物的特征。我们的结果显示,轻度 COVID-19 患者的口咽共生微生物组成明显不同,表现为机会性病原体如 和 的丰度增加, 和 、 和 的丰度减少。基于种水平口咽共生微生物的相对丰度,我们构建了一个微生物分类器,以区分 COVID-19 患者和健康对照者,其中 、 、 和 被鉴定为 COVID-19 组中缺失的最显著特征。几个属的成员,特别是 和 ,在 COVID-19 患者中高度富集,这些患者的严重急性呼吸系统综合征冠状病毒 2(SARS-CoV-2)病毒载量较高,与疾病状态和几个常规临床血液指标显著相关,表明在 COVID-19 患者中,当面临病毒感染的挑战时,几种细菌可能转化为机会性病原体。我们还发现了疾病患者网络中的分类群 和 ,表明这些口咽共生微生物的改变可能通过影响微生物关联模式来影响 COVID-19 的严重程度。总之,这里 SARS-CoV-2 感染患者的样本量(n = 10)较小,因此这些结果只是初步的;然而,我们已经提供了总体特征,即口咽共生微生物的改变和微生物相关性模式与安徽省 COVID-19 的严重程度相关。