Tareen Ammar, Kooshkbaghi Mahdi, Posfai Anna, Ireland William T, McCandlish David M, Kinney Justin B
Simons Center for Quantitative Biology, Cold Spring Harbor Laboratory, Cold Spring Harbor, 11724, NY, USA.
Present Address: Regeneron Pharmaceuticals, Inc., Tarrytown, 10591, NY, USA.
Genome Biol. 2022 Apr 15;23(1):98. doi: 10.1186/s13059-022-02661-7.
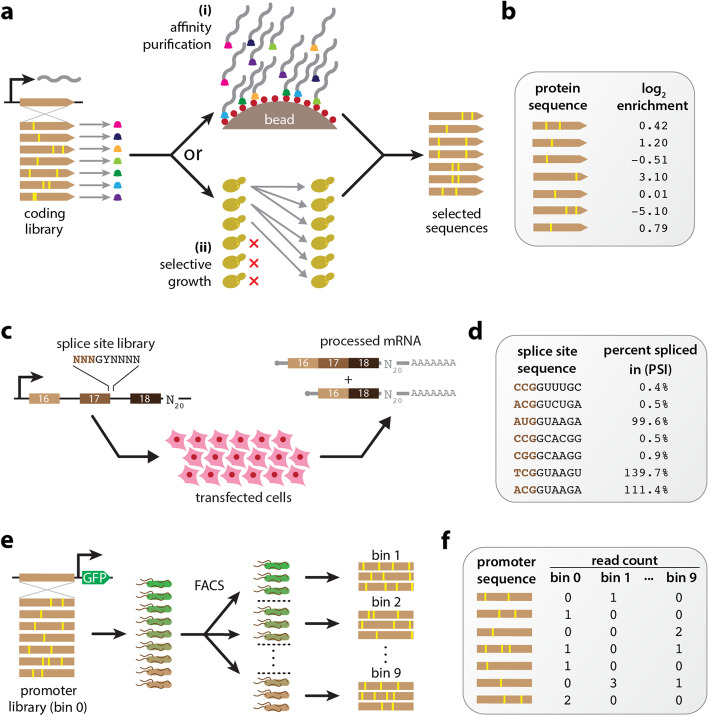
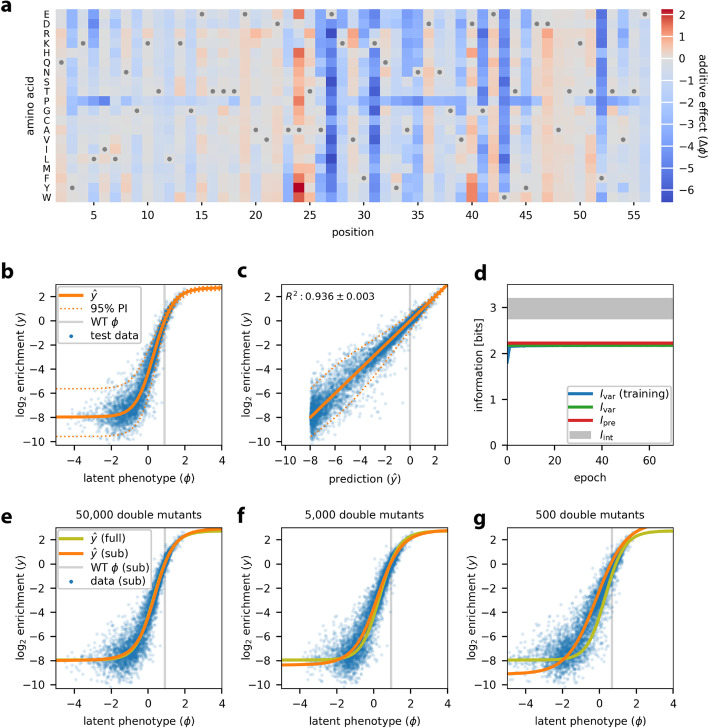
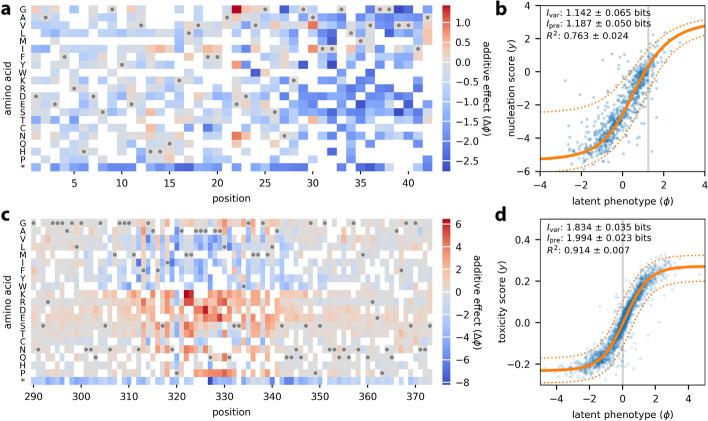
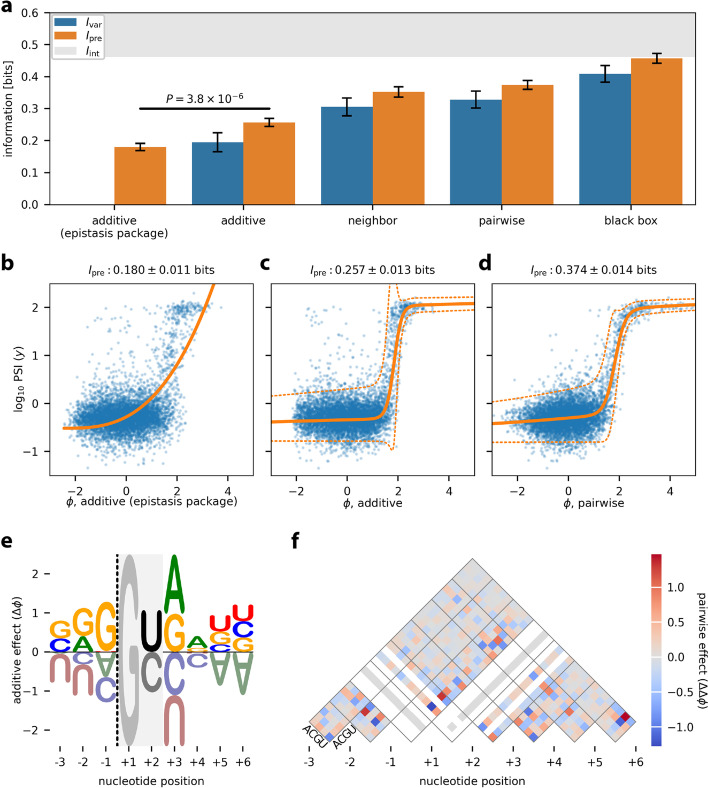
Multiplex assays of variant effect (MAVEs) are a family of methods that includes deep mutational scanning experiments on proteins and massively parallel reporter assays on gene regulatory sequences. Despite their increasing popularity, a general strategy for inferring quantitative models of genotype-phenotype maps from MAVE data is lacking. Here we introduce MAVE-NN, a neural-network-based Python package that implements a broadly applicable information-theoretic framework for learning genotype-phenotype maps-including biophysically interpretable models-from MAVE datasets. We demonstrate MAVE-NN in multiple biological contexts, and highlight the ability of our approach to deconvolve mutational effects from otherwise confounding experimental nonlinearities and noise.
变异效应多重分析(MAVEs)是一类方法,包括对蛋白质的深度突变扫描实验和对基因调控序列的大规模平行报告基因检测。尽管它们越来越受欢迎,但缺乏一种从MAVE数据推断基因型-表型图谱定量模型的通用策略。在这里,我们介绍了MAVE-NN,这是一个基于神经网络的Python软件包,它实现了一个广泛适用的信息论框架,用于从MAVE数据集中学习基因型-表型图谱,包括具有生物物理可解释性的模型。我们在多个生物学背景下展示了MAVE-NN,并强调了我们的方法从其他混淆的实验非线性和噪声中解卷积突变效应的能力。