Institute for Molecular Modeling and Simulation, Department of Material Sciences and Process Engineering, University of Natural Resources and Life Sciences, Vienna, Muthgasse 18, 1190 Vienna, Austria.
Department of Chemical Physics, Institute of Physical Chemistry and Chemical Physics, Faculty of Chemical and Food Technology, Slovak University of Technology in Bratislava, Radlinského 9, 812 37 Bratislava, Slovakia.
J Phys Chem Lett. 2022 May 5;13(17):3812-3818. doi: 10.1021/acs.jpclett.2c00654. Epub 2022 Apr 25.
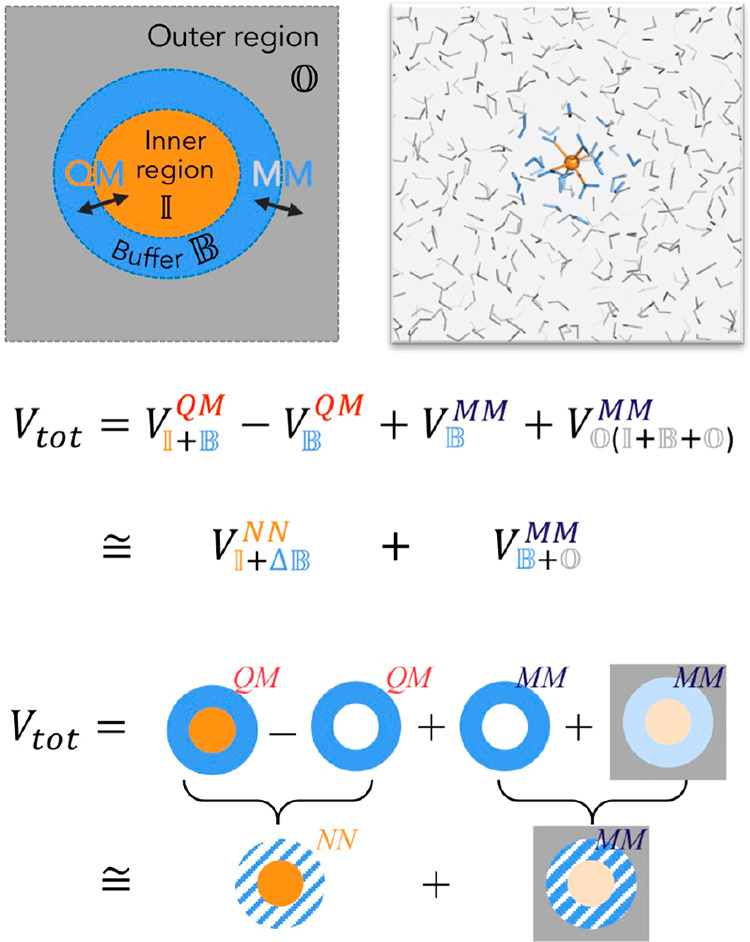
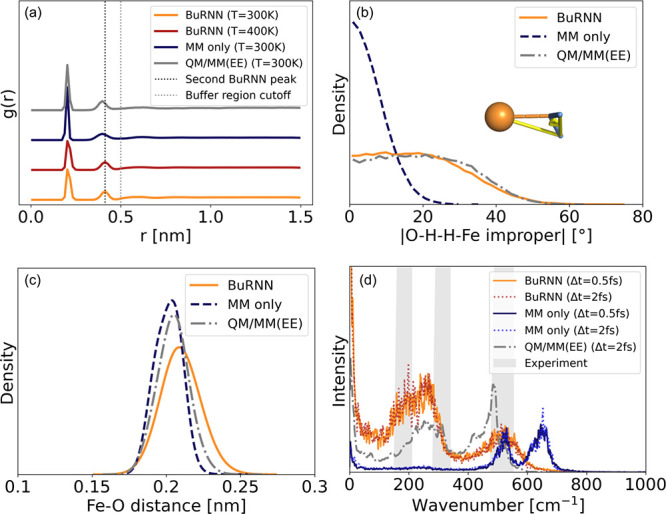
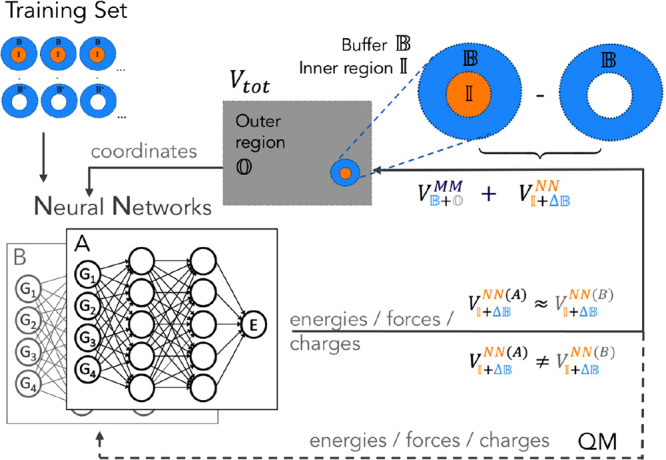
Hybrid quantum mechanics/molecular mechanics (QM/MM) simulations have advanced the field of computational chemistry tremendously. However, they require the partitioning of a system into two different regions that are treated at different levels of theory, which can cause artifacts at the interface. Furthermore, they are still limited by high computational costs of quantum chemical calculations. In this work, we develop the buffer region neural network (BuRNN), an alternative approach to existing QM/MM schemes, which introduces a buffer region that experiences full electronic polarization by the inner QM region to minimize artifacts. The interactions between the QM and the buffer region are described by deep neural networks (NNs), which leads to the high computational efficiency of this hybrid NN/MM scheme while retaining quantum chemical accuracy. We demonstrate the BuRNN approach by performing NN/MM simulations of the hexa-aqua iron complex.
混合量子力学/分子力学(QM/MM)模拟极大地推动了计算化学领域的发展。然而,它们需要将系统划分为两个不同的区域,这些区域分别采用不同的理论水平进行处理,这可能会在界面处产生伪影。此外,它们仍然受到量子化学计算高计算成本的限制。在这项工作中,我们开发了缓冲区域神经网络(BuRNN),这是一种替代现有 QM/MM 方案的方法,它引入了一个缓冲区域,该区域通过内部 QM 区域经历完全的电子极化,以最小化伪影。QM 和缓冲区域之间的相互作用由深度神经网络(NN)描述,这导致这种混合 NN/MM 方案具有很高的计算效率,同时保持了量子化学精度。我们通过对六水合铁配合物进行 NN/MM 模拟来演示 BuRNN 方法。