Karwounopoulos Johannes, Wu Zhiyi, Tkaczyk Sara, Wang Shuzhe, Baskerville Adam, Ranasinghe Kavindri, Langer Thierry, Wood Geoffrey P F, Wieder Marcus, Boresch Stefan
Faculty of Chemistry, Institute of Computational Biological Chemistry, University Vienna, Währingerstr. 17, 1090 Vienna, Austria.
Vienna Doctoral School of Chemistry (DoSChem), University of Vienna, Währingerstr. 42, 1090 Vienna, Austria.
J Phys Chem B. 2024 Jul 18;128(28):6693-6703. doi: 10.1021/acs.jpcb.4c01417. Epub 2024 Jul 8.
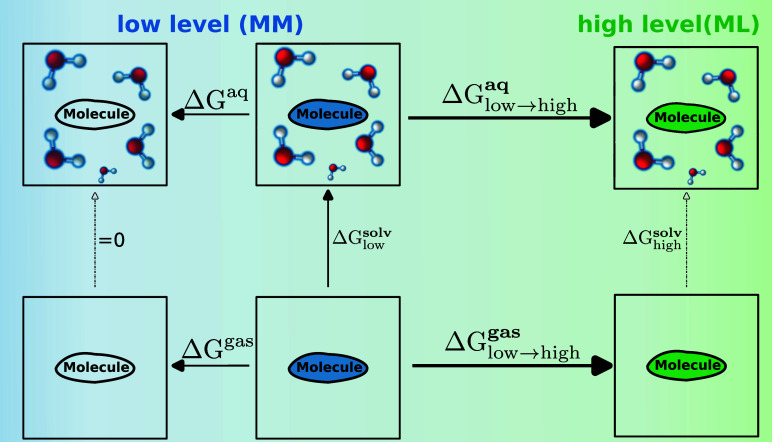
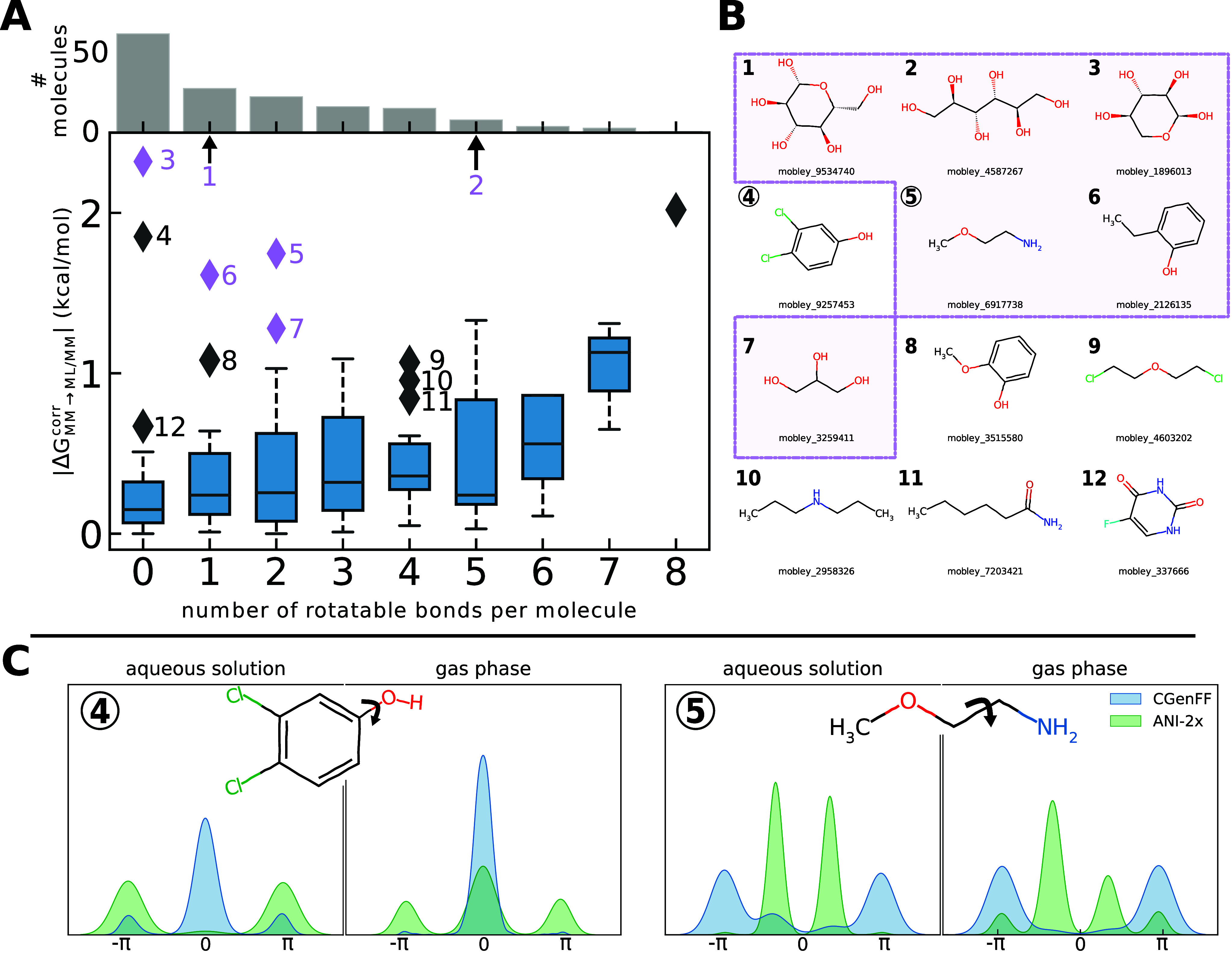
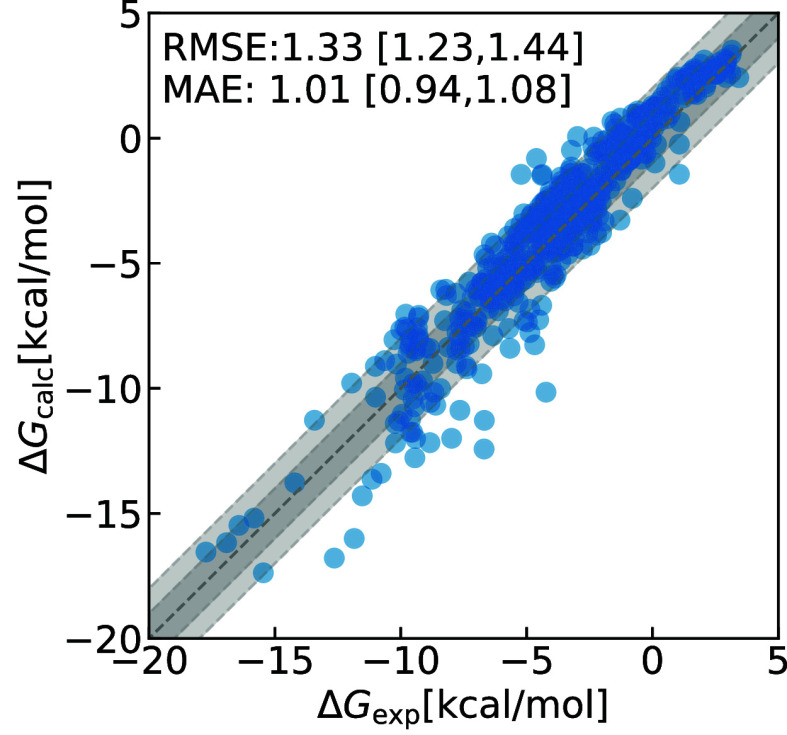
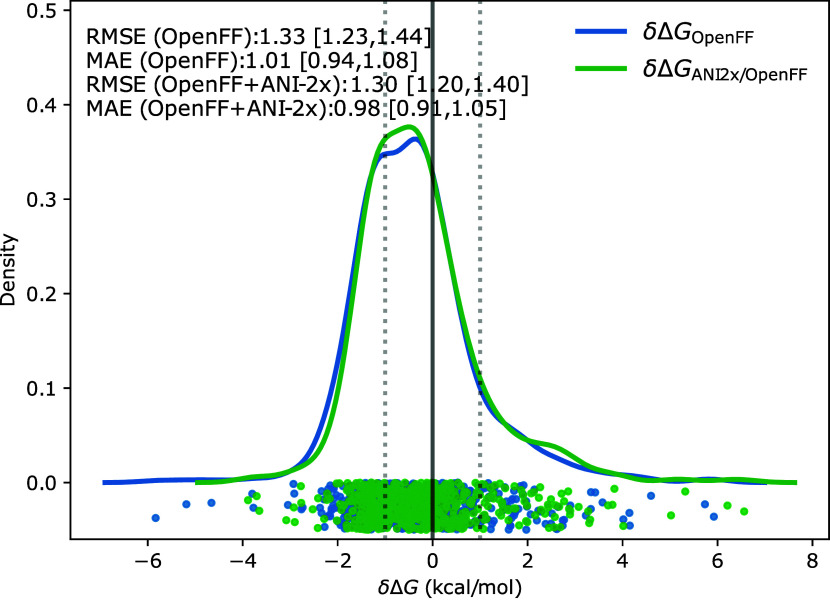
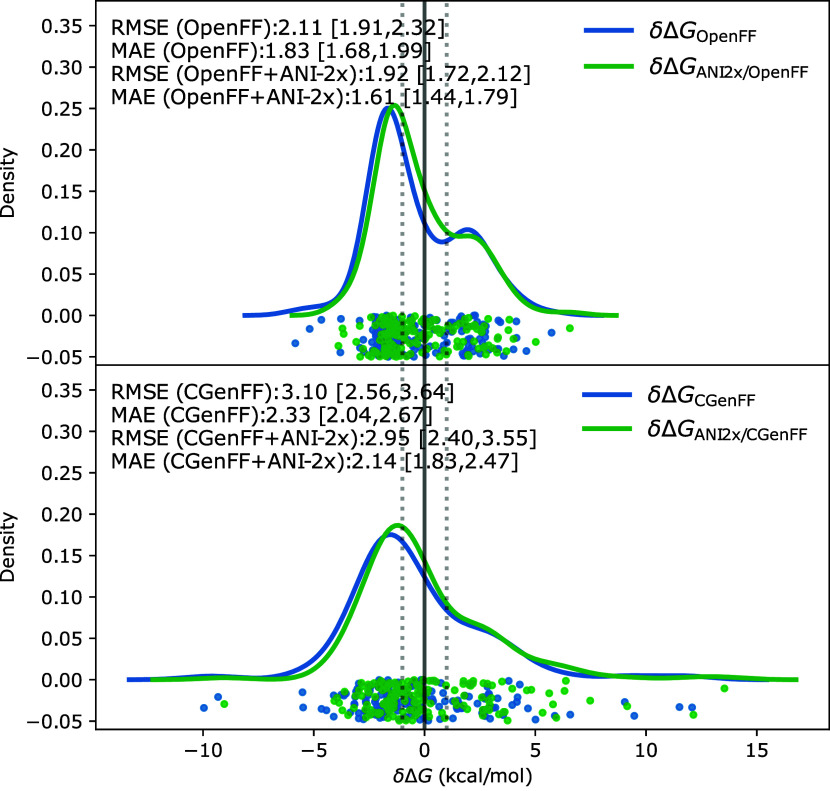
We present a comprehensive study investigating the potential gain in accuracy for calculating absolute solvation free energies (ASFE) using a neural network potential to describe the intramolecular energy of the solute. We calculated the ASFE for most compounds from the FreeSolv database using the Open Force Field (OpenFF) and compared them to earlier results obtained with the CHARMM General Force Field (CGenFF). By applying a nonequilibrium (NEQ) switching approach between the molecular mechanics (MM) description (either OpenFF or CGenFF) and the neural net potential (NNP)/MM level of theory (using ANI-2x as the NNP potential), we attempted to improve the accuracy of the calculated ASFEs. The predictive performance of the results did not change when this approach was applied to all 589 small molecules in the FreeSolv database that ANI-2x can describe. When selecting a subset of 156 molecules, focusing on compounds where the force fields performed poorly, we saw a slight improvement in the root-mean-square error (RMSE) and mean absolute error (MAE). The majority of our calculations utilized unidirectional NEQ protocols based on Jarzynski's equation. Additionally, we conducted bidirectional NEQ switching for a subset of 156 solutes. Notably, only a small fraction (10 out of 156) exhibited statistically significant discrepancies between unidirectional and bidirectional NEQ switching free energy estimates.
我们开展了一项全面研究,探究使用神经网络势描述溶质分子内能量来计算绝对溶剂化自由能(ASFE)时在精度方面的潜在提升。我们使用开放力场(OpenFF)计算了FreeSolv数据库中大多数化合物的ASFE,并将其与使用CHARMM通用力场(CGenFF)获得的早期结果进行比较。通过在分子力学(MM)描述(OpenFF或CGenFF)与神经网络势(NNP)/MM理论水平(使用ANI - 2x作为NNP势)之间应用非平衡(NEQ)切换方法,我们试图提高计算得到的ASFE的精度。当将此方法应用于FreeSolv数据库中ANI - 2x能够描述的所有589个小分子时,结果的预测性能并未改变。当选择156个分子的子集,重点关注力场表现不佳的化合物时,我们看到均方根误差(RMSE)和平均绝对误差(MAE)略有改善。我们的大多数计算使用了基于雅尔津斯基方程的单向NEQ协议。此外,我们对156个溶质的子集进行了双向NEQ切换。值得注意的是,在单向和双向NEQ切换自由能估计之间,只有一小部分(156个中的10个)表现出统计学上的显著差异。