Lie Pearl P Y, Yoo Lang, Goulbourne Chris N, Berg Martin J, Stavrides Philip, Huo Chunfeng, Lee Ju-Hyun, Nixon Ralph A
Center for Dementia Research, Nathan S. Kline Institute, Orangeburg, NY 10962, USA.
Department of Psychiatry, New York University Langone Medical Center, New York, NY 10016, USA.
Sci Adv. 2022 Apr 29;8(17):eabj5716. doi: 10.1126/sciadv.abj5716.
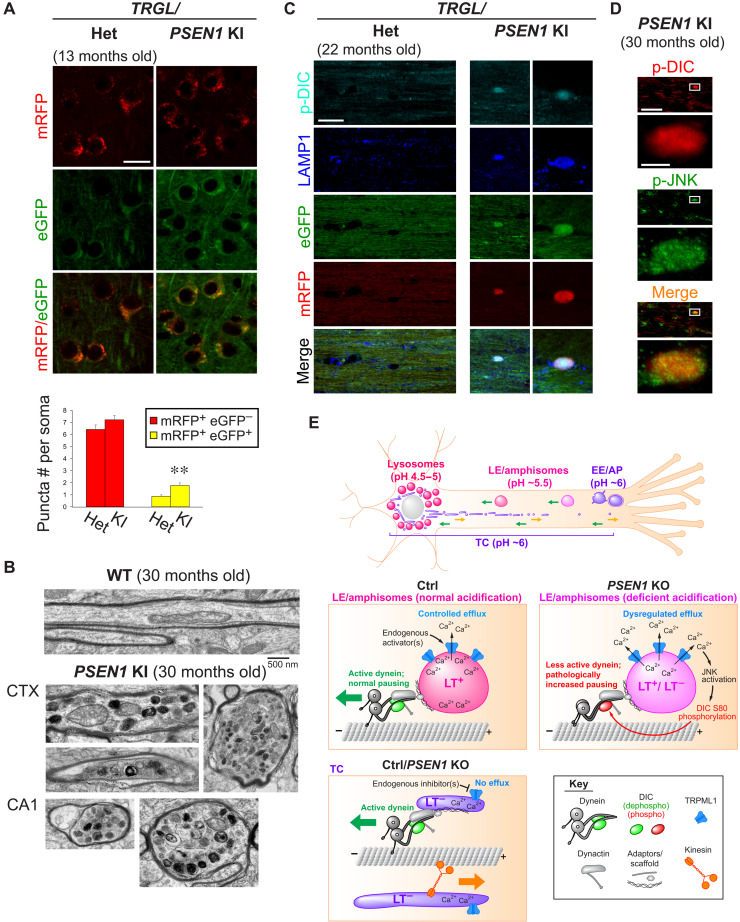
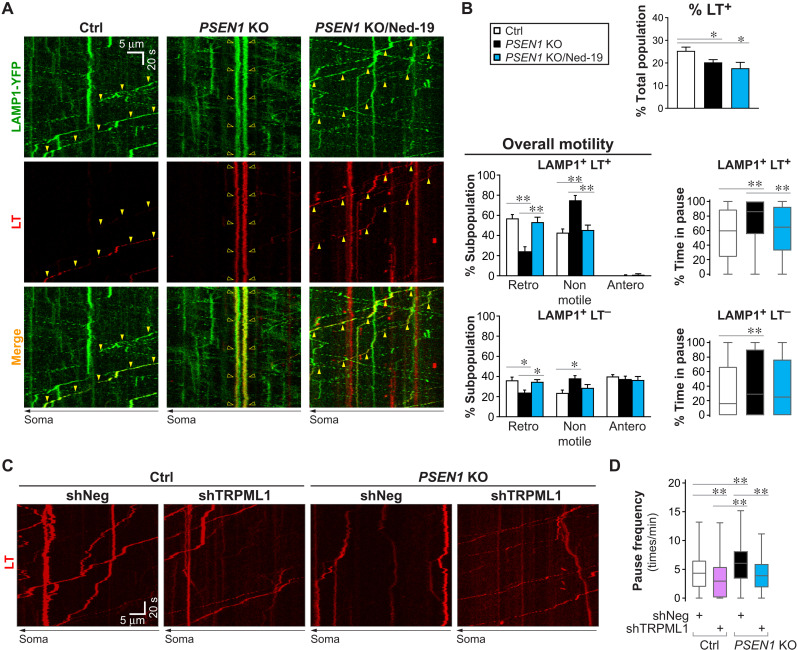
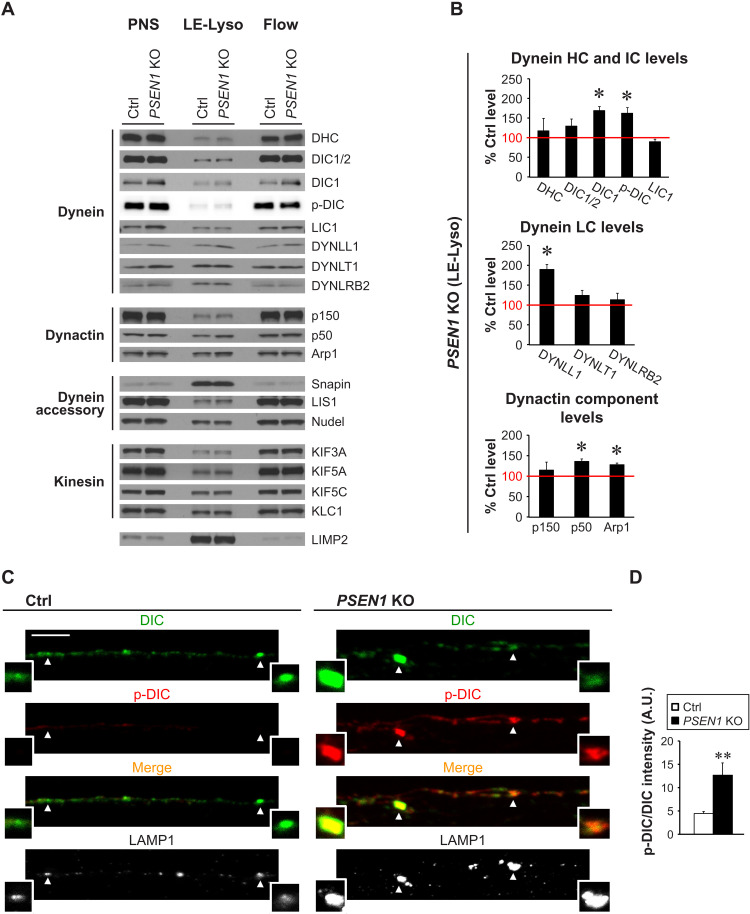
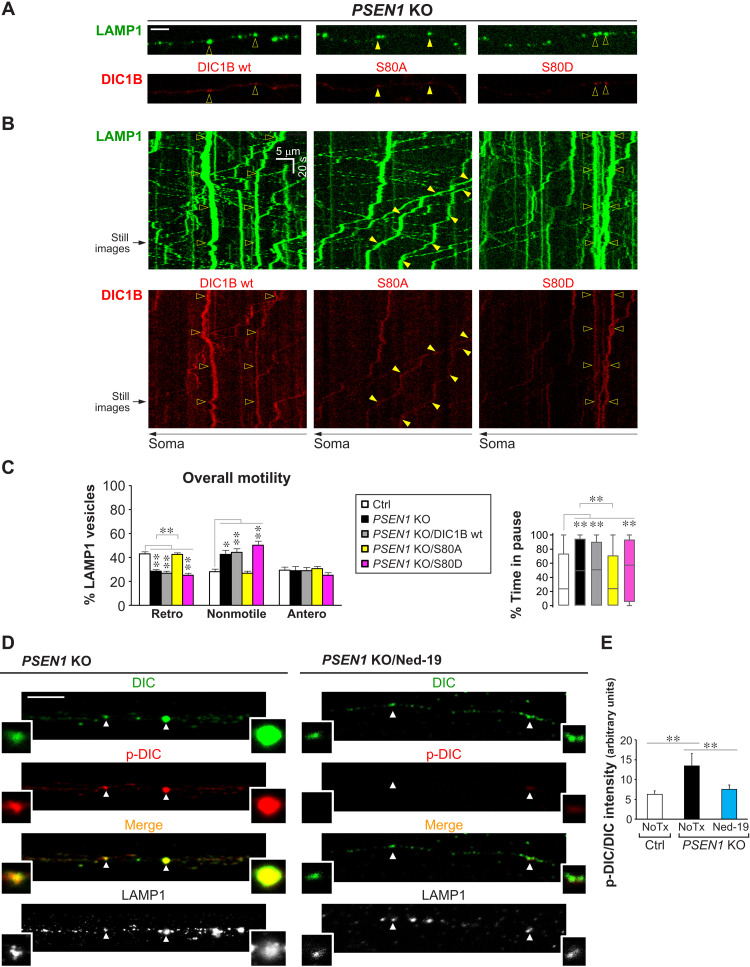
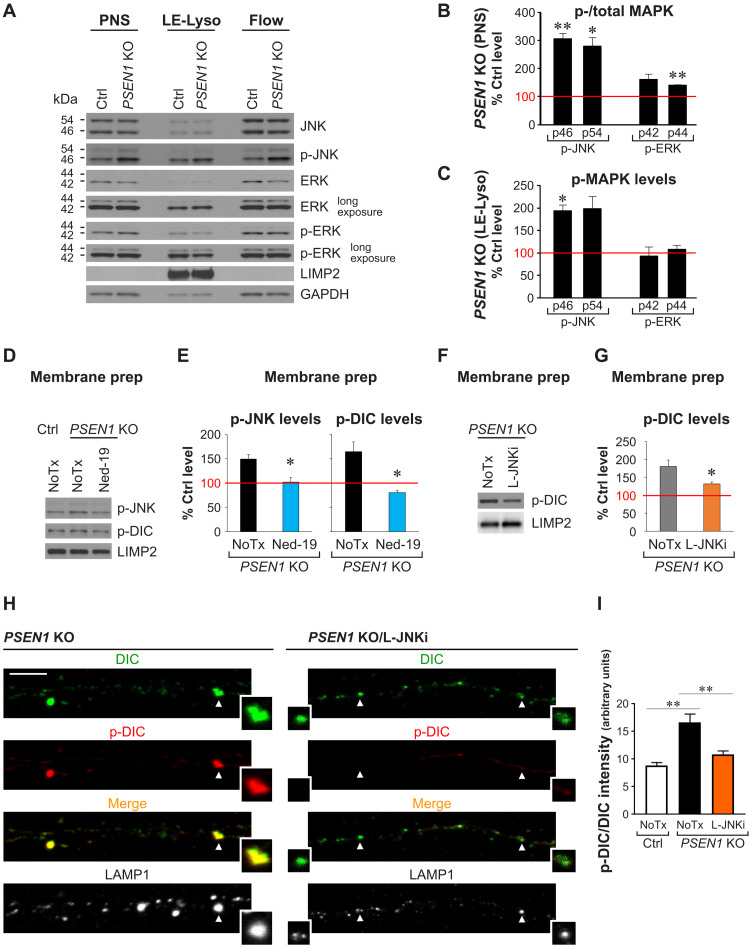
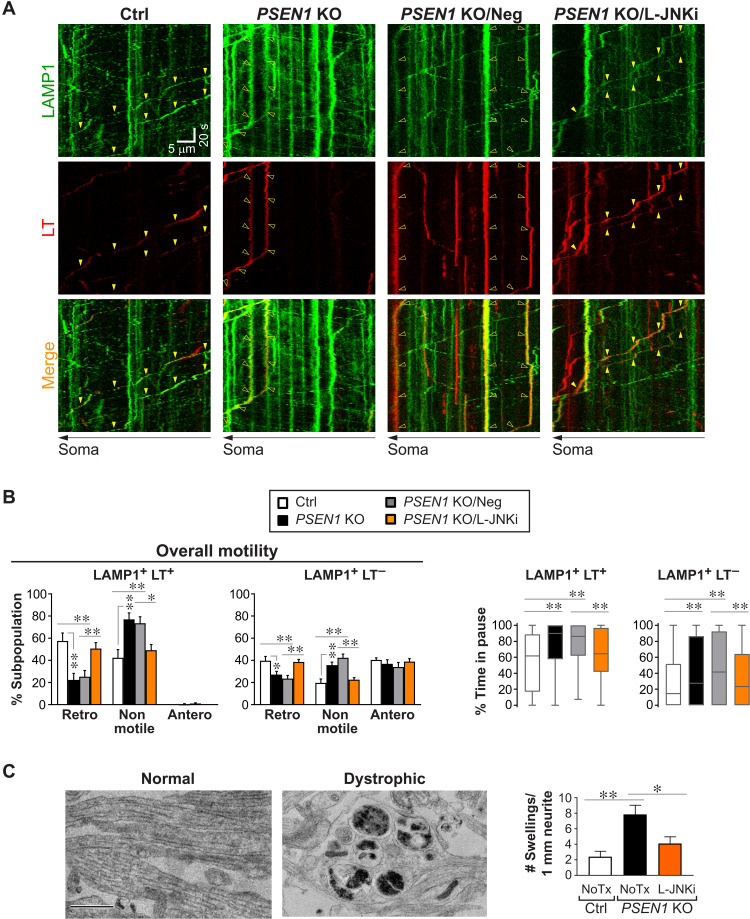
Dysfunction and mistrafficking of organelles in autophagy- and endosomal-lysosomal pathways are implicated in neurodegenerative diseases. Here, we reveal selective vulnerability of maturing degradative organelles (late endosomes/amphisomes) to disease-relevant local calcium dysregulation. These organelles undergo exclusive retrograde transport in axons, with occasional pauses triggered by regulated calcium efflux from agonist-evoked transient receptor potential cation channel mucolipin subfamily member 1 (TRPML1) channels-an effect greatly exaggerated by exogenous agonist mucolipin synthetic agonist 1 (ML-SA1). Deacidification of degradative organelles, as seen after Presenilin 1 (PSEN1) loss of function, induced pathological constitutive "inside-out" TRPML1 hyperactivation, slowing their transport comparably to ML-SA1 and causing accumulation in dystrophic axons. The mechanism involved calcium-mediated c-Jun N-terminal kinase (JNK) activation, which hyperphosphorylated dynein intermediate chain (DIC), reducing dynein activity. Blocking TRPML1 activation, JNK activity, or DIC1B serine-80 phosphorylation reversed transport deficits in knockout neurons. Our results, including features demonstrated in Alzheimer-mutant knockin mice, define a mechanism linking dysfunction and mistrafficking in lysosomal pathways to neuritic dystrophy under neurodegenerative conditions.
自噬和内体-溶酶体途径中细胞器的功能障碍和错误运输与神经退行性疾病有关。在这里,我们揭示了成熟的降解细胞器(晚期内体/两性体)对疾病相关的局部钙失调具有选择性易损性。这些细胞器在轴突中进行独特的逆行运输,偶尔会因激动剂诱发的瞬时受体电位阳离子通道黏脂蛋白亚家族成员1(TRPML1)通道的钙外流调节而暂停——外源性激动剂黏脂蛋白合成激动剂1(ML-SA1)会大大增强这种效应。如早老素1(PSEN1)功能丧失后所见,降解细胞器的去酸化诱导了病理性的组成性“由内而外”的TRPML1过度激活,使其运输速度与ML-SA1相当减慢,并导致在营养不良的轴突中积累。其机制涉及钙介导的c-Jun氨基末端激酶(JNK)激活,后者使动力蛋白中间链(DIC)过度磷酸化,降低动力蛋白活性。阻断TRPML1激活、JNK活性或DIC1B丝氨酸80磷酸化可逆转基因敲除神经元中的运输缺陷。我们的结果,包括在阿尔茨海默病突变基因敲入小鼠中所展示的特征,确定了一种在神经退行性疾病条件下将溶酶体途径中的功能障碍和错误运输与神经突营养不良联系起来的机制。