Levina Elena O, Chernyshov Ivan Y, Voronin Alexander P, Alekseiko Leonid N, Stash Adam I, Vener Mikhail V
Moscow Institute of Physics and Technology Dolgoprudny Russia.
Research Centre of Biotechnology, Russian Academy of Sciences Moscow Russia.
RSC Adv. 2019 Apr 23;9(22):12520-12537. doi: 10.1039/c9ra02116g. eCollection 2019 Apr 17.
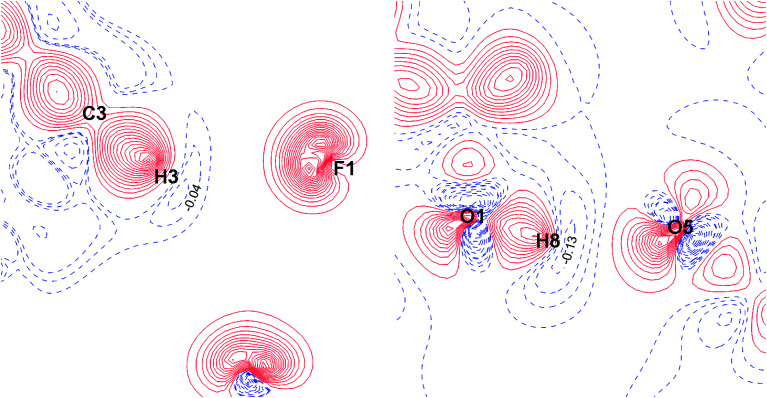
The nature and strength of weak interactions with organic fluorine in the solid state are revealed by periodic density functional theory (periodic DFT) calculations coupled with experimental data on the structure and sublimation thermodynamics of crystalline organofluorine compounds. To minimize other intermolecular interactions, several sets of crystals of perfluorinated and partially fluorinated organic molecules are considered. This allows us to establish the theoretical levels providing an adequate description of the metric and electron-density parameters of the C-F⋯F-C interactions and the sublimation enthalpy of crystalline perfluorinated compounds. A detailed comparison of the C-F⋯F-C and C-H⋯F-C interactions is performed using the relaxed molecular geometry in the studied crystals. The change in the crystalline packing of aromatic compounds during their partial fluorination points to the structure-directing role of C-H⋯F-C interactions due to the dominant electrostatic contribution to these contacts. C-H⋯F-C and C-H⋯O interactions are found to be identical in nature and comparable in energy. The factors that determine the contribution of these interactions to the crystal packing are revealed. The reliability of the results is confirmed by considering the superposition of the electrostatic potential and electron density gradient fields in the area of the investigated intermolecular interactions.
通过周期性密度泛函理论(周期性DFT)计算,并结合结晶有机氟化合物的结构和升华热力学实验数据,揭示了固态下与有机氟的弱相互作用的性质和强度。为了最小化其他分子间相互作用,考虑了几组全氟和部分氟化有机分子的晶体。这使我们能够确定理论水平,从而充分描述C-F⋯F-C相互作用的度量和电子密度参数以及结晶全氟化合物的升华焓。利用所研究晶体中松弛的分子几何结构,对C-F⋯F-C和C-H⋯F-C相互作用进行了详细比较。芳香族化合物在部分氟化过程中晶体堆积的变化表明,由于这些接触中主要的静电贡献,C-H⋯F-C相互作用具有结构导向作用。发现C-H⋯F-C和C-H⋯O相互作用在性质上相同且能量相当。揭示了决定这些相互作用对晶体堆积贡献的因素。通过考虑所研究分子间相互作用区域中静电势和电子密度梯度场的叠加,证实了结果的可靠性。