Jiang Mingjian, Li Zhen, Zhang Shugang, Wang Shuang, Wang Xiaofeng, Yuan Qing, Wei Zhiqiang
Department of Computer Science and Technology, Ocean University of China China
RSC Adv. 2020 Jun 1;10(35):20701-20712. doi: 10.1039/d0ra02297g. eCollection 2020 May 27.
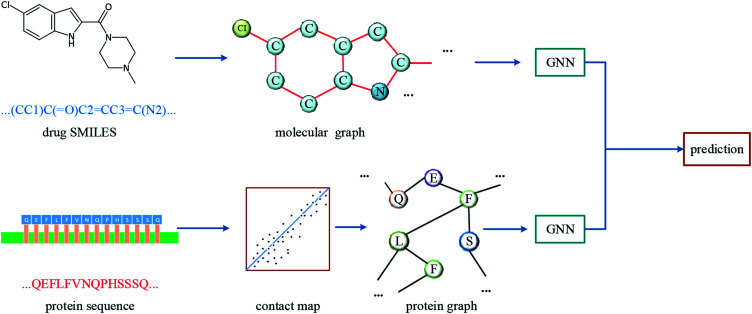
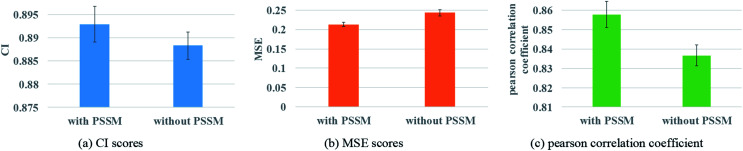
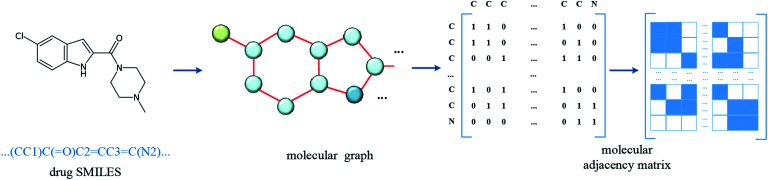
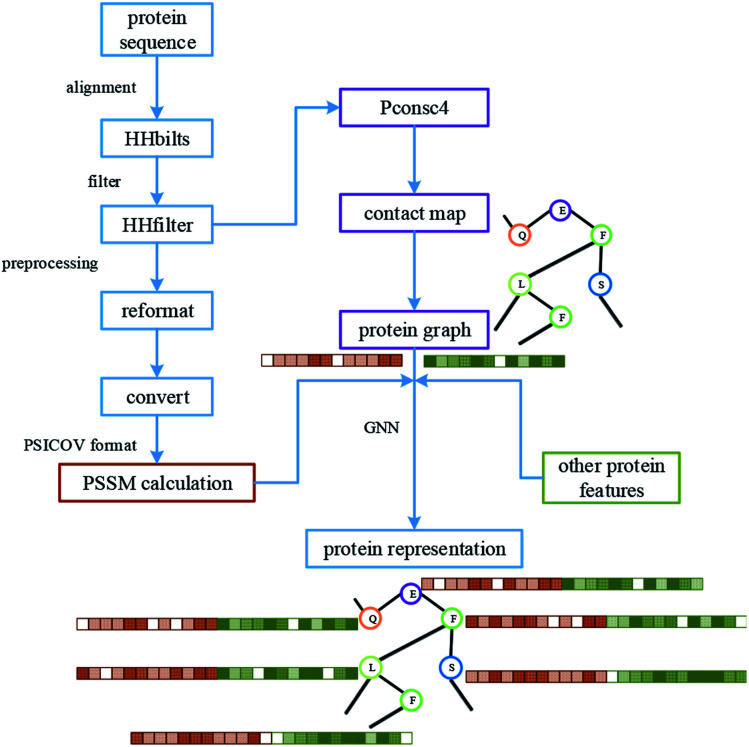
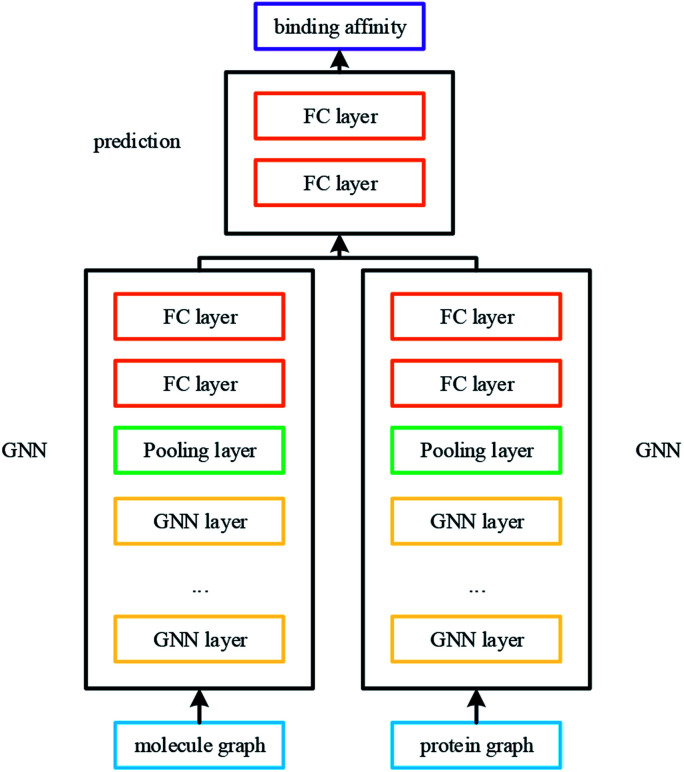
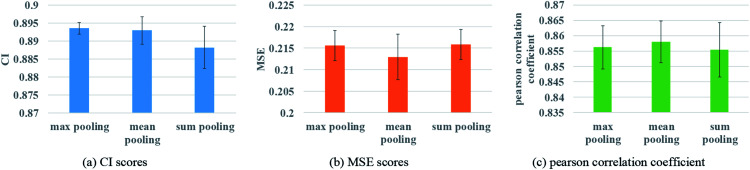
Computer-aided drug design uses high-performance computers to simulate the tasks in drug design, which is a promising research area. Drug-target affinity (DTA) prediction is the most important step of computer-aided drug design, which could speed up drug development and reduce resource consumption. With the development of deep learning, the introduction of deep learning to DTA prediction and improving the accuracy have become a focus of research. In this paper, utilizing the structural information of molecules and proteins, two graphs of drug molecules and proteins are built up respectively. Graph neural networks are introduced to obtain their representations, and a method called DGraphDTA is proposed for DTA prediction. Specifically, the protein graph is constructed based on the contact map output from the prediction method, which could predict the structural characteristics of the protein according to its sequence. It can be seen from the test of various metrics on benchmark datasets that the method proposed in this paper has strong robustness and generalizability.
计算机辅助药物设计利用高性能计算机来模拟药物设计中的任务,这是一个很有前景的研究领域。药物-靶点亲和力(DTA)预测是计算机辅助药物设计中最重要的步骤,它可以加速药物开发并减少资源消耗。随着深度学习的发展,将深度学习引入DTA预测并提高其准确性已成为研究的一个重点。在本文中,利用分子和蛋白质的结构信息,分别构建了药物分子和蛋白质的两个图。引入图神经网络以获得它们的表示,并提出了一种名为DGraphDTA的方法用于DTA预测。具体来说,蛋白质图是基于预测方法输出的接触图构建的,该方法可以根据蛋白质序列预测其结构特征。从在基准数据集上的各种指标测试中可以看出,本文提出的方法具有很强的鲁棒性和通用性。