Committee on Genetics, Genomics, & Systems Biology, University of Chicago, Chicago, Illinois, United States of America.
Department of Human Genetics, University of Chicago, Chicago, Illinois, United States of America.
PLoS Genet. 2022 May 6;18(5):e1010170. doi: 10.1371/journal.pgen.1010170. eCollection 2022 May.
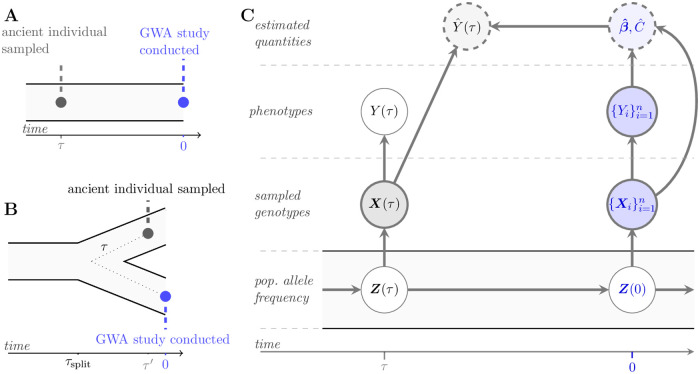
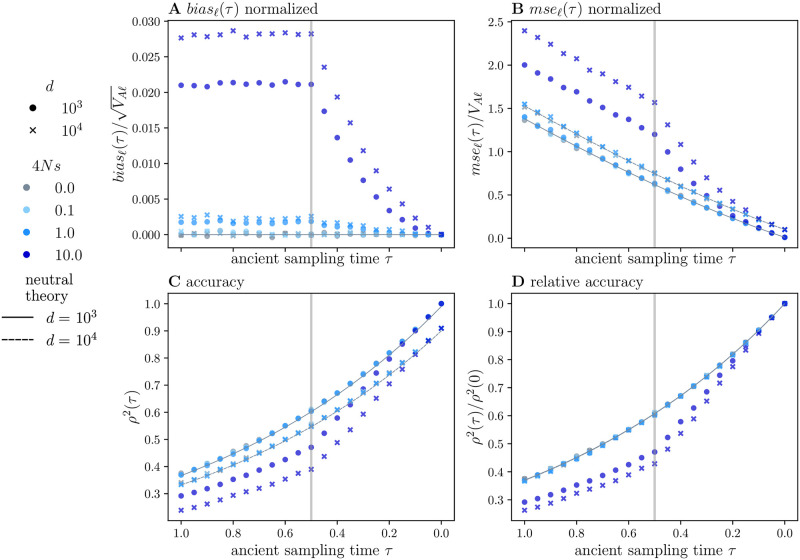
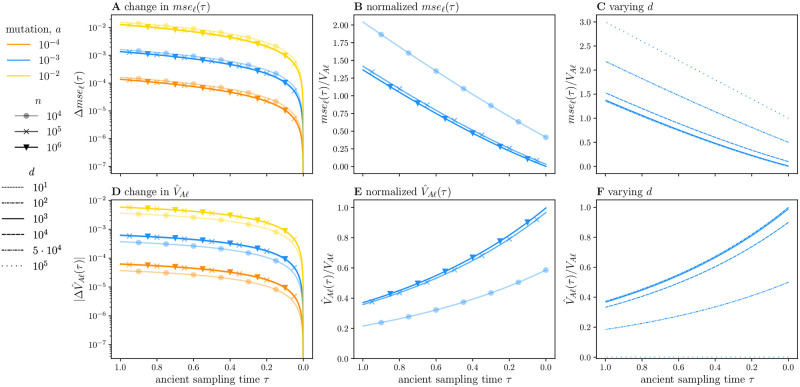
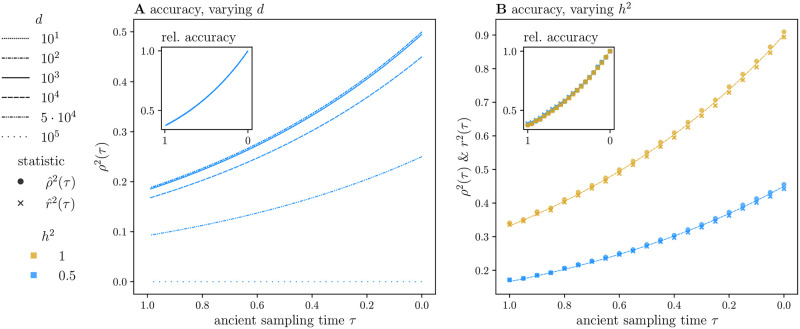
Polygenic scores link the genotypes of ancient individuals to their phenotypes, which are often unobservable, offering a tantalizing opportunity to reconstruct complex trait evolution. In practice, however, interpretation of ancient polygenic scores is subject to numerous assumptions. For one, the genome-wide association (GWA) studies from which polygenic scores are derived, can only estimate effect sizes for loci segregating in contemporary populations. Therefore, a GWA study may not correctly identify all loci relevant to trait variation in the ancient population. In addition, the frequencies of trait-associated loci may have changed in the intervening years. Here, we devise a theoretical framework to quantify the effect of this allelic turnover on the statistical properties of polygenic scores as functions of population genetic dynamics, trait architecture, power to detect significant loci, and the age of the ancient sample. We model the allele frequencies of loci underlying trait variation using the Wright-Fisher diffusion, and employ the spectral representation of its transition density to find analytical expressions for several error metrics, including the expected sample correlation between the polygenic scores of ancient individuals and their true phenotypes, referred to as polygenic score accuracy. Our theory also applies to a two-population scenario and demonstrates that allelic turnover alone may explain a substantial percentage of the reduced accuracy observed in cross-population predictions, akin to those performed in human genetics. Finally, we use simulations to explore the effects of recent directional selection, a bias-inducing process, on the statistics of interest. We find that even in the presence of bias, weak selection induces minimal deviations from our neutral expectations for the decay of polygenic score accuracy. By quantifying the limitations of polygenic scores in an explicit evolutionary context, our work lays the foundation for the development of more sophisticated statistical procedures to analyze both temporally and geographically resolved polygenic scores.
多基因分数将古代个体的基因型与其表型联系起来,而这些表型通常是不可观察的,这为重建复杂性状进化提供了诱人的机会。然而,在实践中,对古代多基因分数的解释受到许多假设的限制。首先,多基因分数所源自的全基因组关联(GWA)研究只能估计在当代人群中分离的基因座的效应大小。因此,GWA 研究可能无法正确识别与古代人群中性状变异相关的所有基因座。此外,性状相关基因座的频率可能在 intervening 年中发生了变化。在这里,我们设计了一个理论框架,以量化等位基因转换对多基因分数统计性质的影响,这些影响是作为群体遗传动态、性状结构、检测显著基因座的能力以及古代样本年龄的函数。我们使用 Wright-Fisher 扩散来模拟性状变异下的基因座等位基因频率,并利用其转移密度的谱表示来找到几个误差度量的解析表达式,包括古代个体的多基因分数与真实表型之间的期望样本相关,称为多基因分数准确性。我们的理论也适用于两群体情况,并表明仅等位基因转换就可能解释跨群体预测中观察到的准确性降低的很大一部分,类似于人类遗传学中的预测。最后,我们使用模拟来探索近期定向选择(一种产生偏差的过程)对感兴趣的统计数据的影响。我们发现,即使存在偏差,弱选择也会使多基因分数准确性衰减的偏离我们的中性预期最小。通过在明确的进化背景下量化多基因分数的局限性,我们的工作为分析时间和地理上分辨率的多基因分数的更复杂的统计程序的发展奠定了基础。