Department of Proteomics and Signal Transduction, Max Planck Institute of Biochemistry, Martinsried, Germany.
Proteomics Program, NNF Center for Protein Research, Faculty of Health Sciences, University of Copenhagen, Copenhagen, Denmark.
PLoS Biol. 2022 May 16;20(5):e3001636. doi: 10.1371/journal.pbio.3001636. eCollection 2022 May.
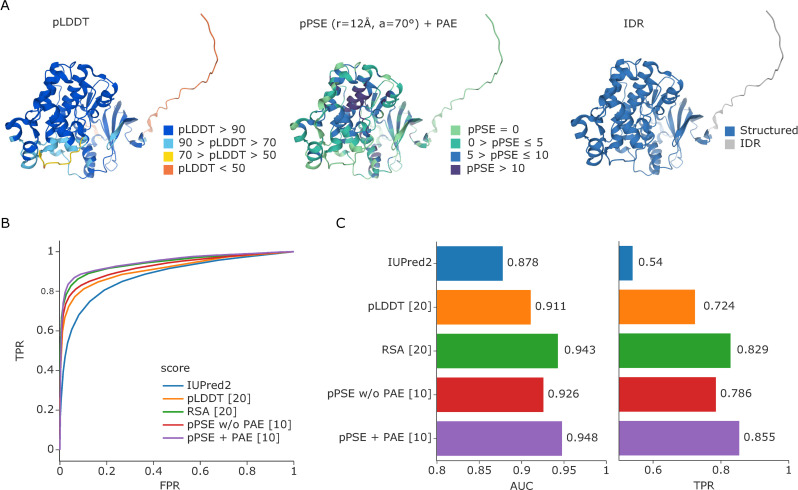
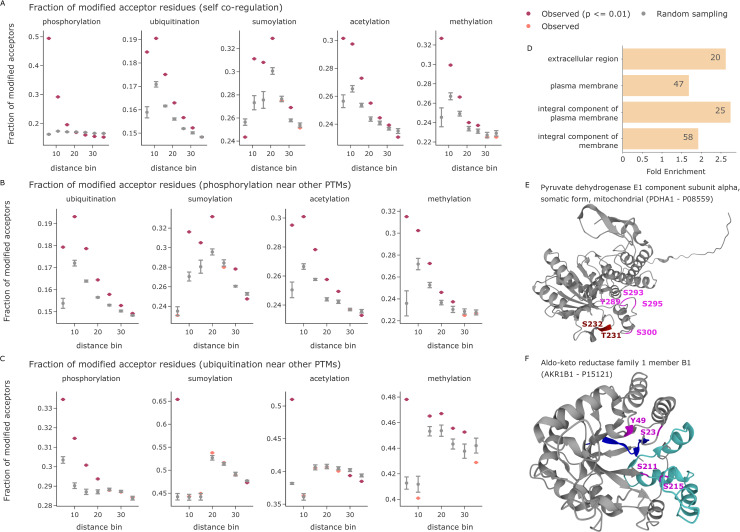
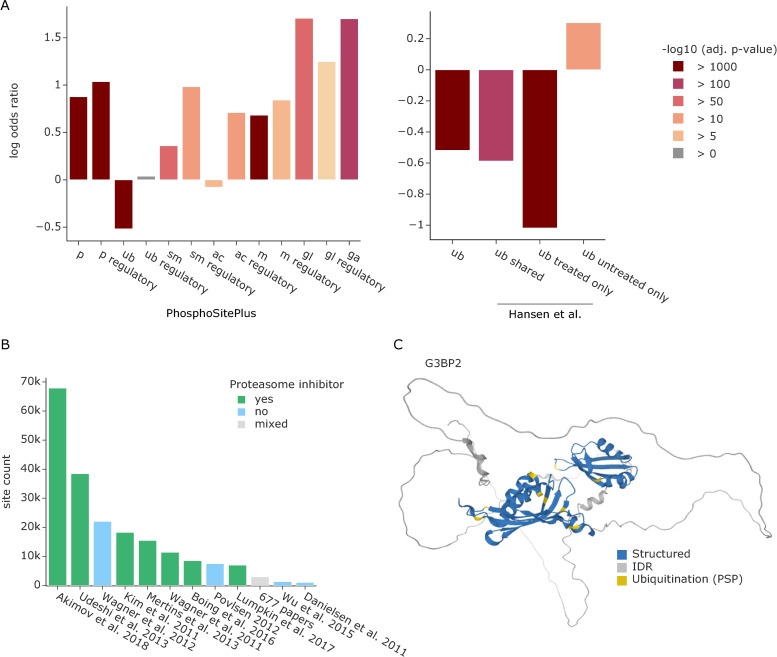
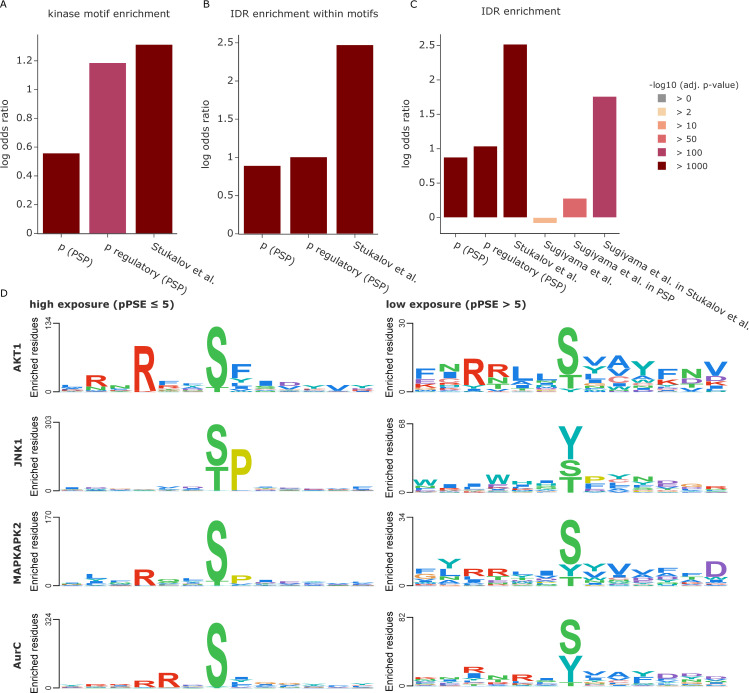
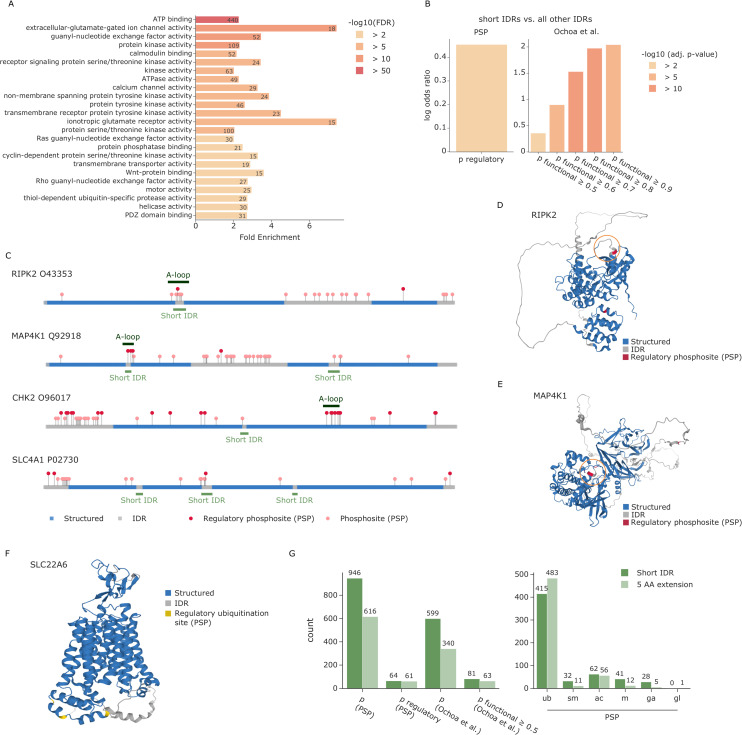
The recent revolution in computational protein structure prediction provides folding models for entire proteomes, which can now be integrated with large-scale experimental data. Mass spectrometry (MS)-based proteomics has identified and quantified tens of thousands of posttranslational modifications (PTMs), most of them of uncertain functional relevance. In this study, we determine the structural context of these PTMs and investigate how this information can be leveraged to pinpoint potential regulatory sites. Our analysis uncovers global patterns of PTM occurrence across folded and intrinsically disordered regions. We found that this information can help to distinguish regulatory PTMs from those marking improperly folded proteins. Interestingly, the human proteome contains thousands of proteins that have large folded domains linked by short, disordered regions that are strongly enriched in regulatory phosphosites. These include well-known kinase activation loops that induce protein conformational changes upon phosphorylation. This regulatory mechanism appears to be widespread in kinases but also occurs in other protein families such as solute carriers. It is not limited to phosphorylation but includes ubiquitination and acetylation sites as well. Furthermore, we performed three-dimensional proximity analysis, which revealed examples of spatial coregulation of different PTM types and potential PTM crosstalk. To enable the community to build upon these first analyses, we provide tools for 3D visualization of proteomics data and PTMs as well as python libraries for data accession and processing.
最近的计算蛋白质结构预测革命为整个蛋白质组提供了折叠模型,这些模型现在可以与大规模的实验数据集成。基于质谱(MS)的蛋白质组学已经鉴定和定量了成千上万的翻译后修饰(PTM),其中大多数具有不确定的功能相关性。在这项研究中,我们确定了这些 PTM 的结构上下文,并研究了如何利用这些信息来确定潜在的调节位点。我们的分析揭示了折叠区和固有无序区中 PTM 发生的全局模式。我们发现,这些信息可以帮助区分调节 PTM 和标记错误折叠蛋白的 PTM。有趣的是,人类蛋白质组中包含数千种蛋白质,它们的大折叠结构域由短的、无序的区域连接,这些区域富含调节磷酸化位点。其中包括众所周知的激酶激活环,在磷酸化后诱导蛋白质构象变化。这种调节机制似乎在激酶中广泛存在,但也存在于其他蛋白质家族,如溶质载体。它不仅限于磷酸化,还包括泛素化和乙酰化位点。此外,我们进行了三维接近分析,揭示了不同 PTM 类型和潜在 PTM 串扰的空间共调节的例子。为了使社区能够在此基础上进行这些首次分析,我们提供了用于蛋白质组学数据和 PTM 的三维可视化工具,以及用于数据访问和处理的 python 库。