Division of Dermatology, Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, California.
Department of Molecular and Medical Pharmacology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, California.
Cancer Discov. 2022 Aug 5;12(8):1942-1959. doi: 10.1158/2159-8290.CD-21-1463.
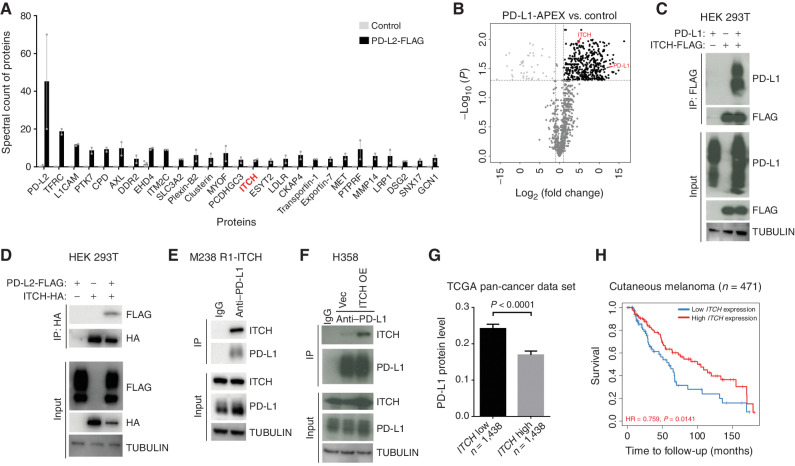
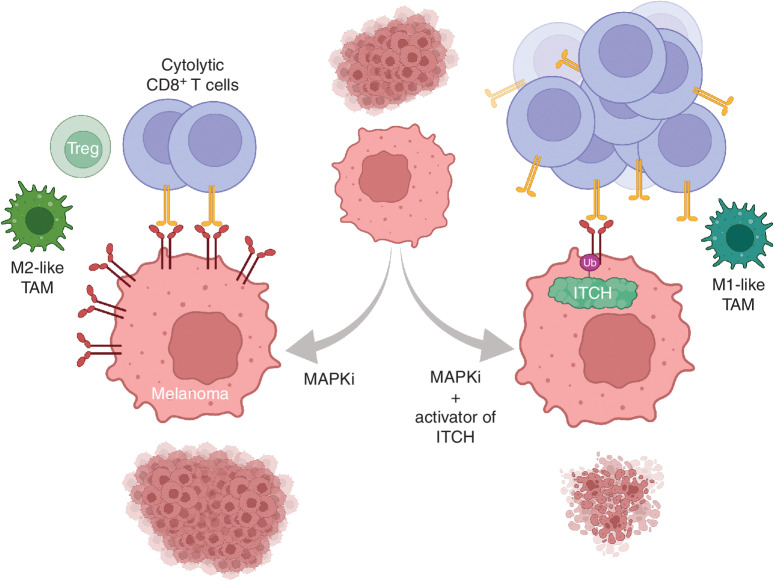
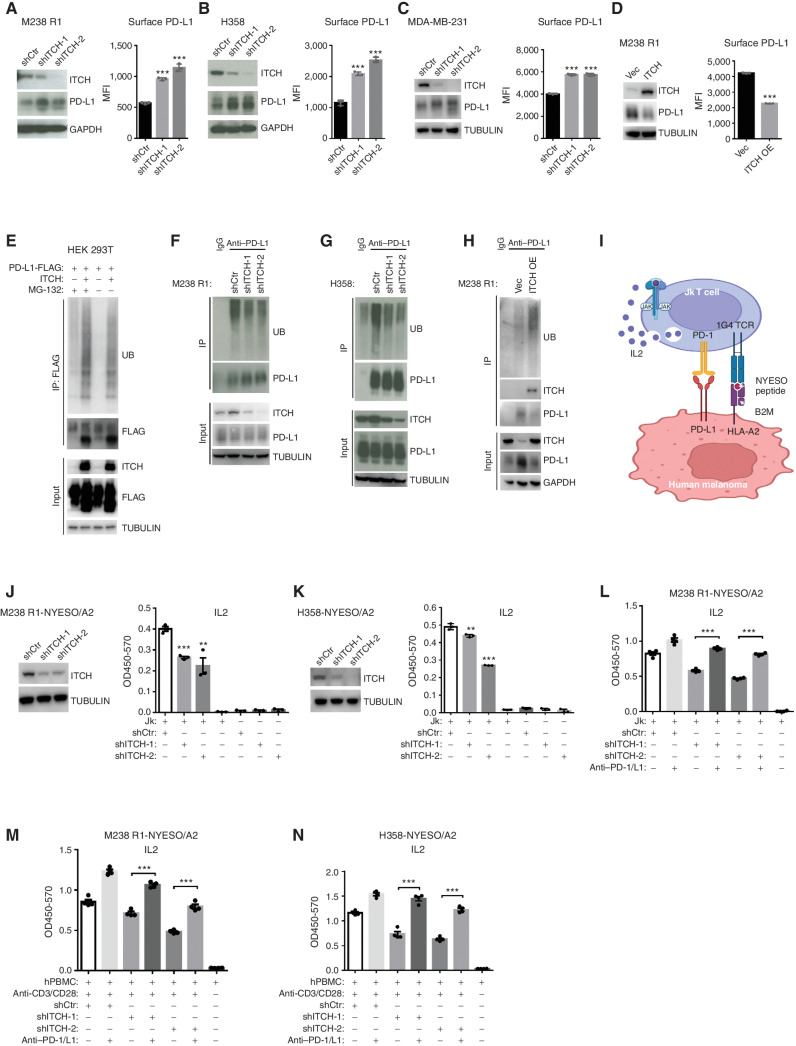
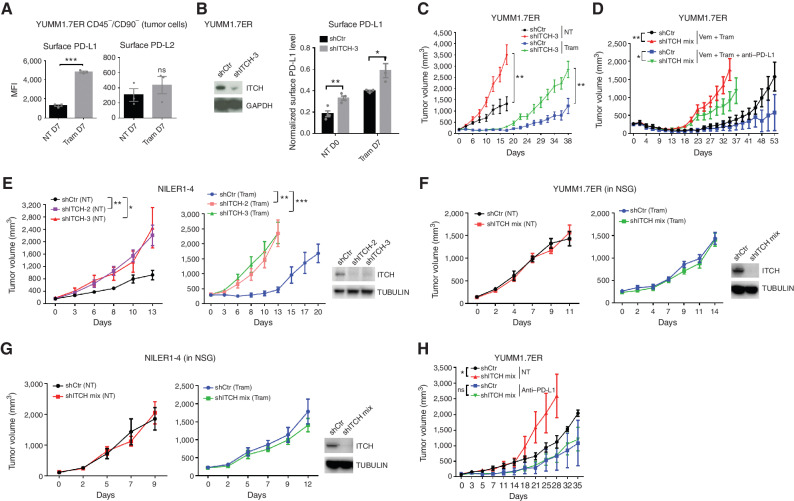
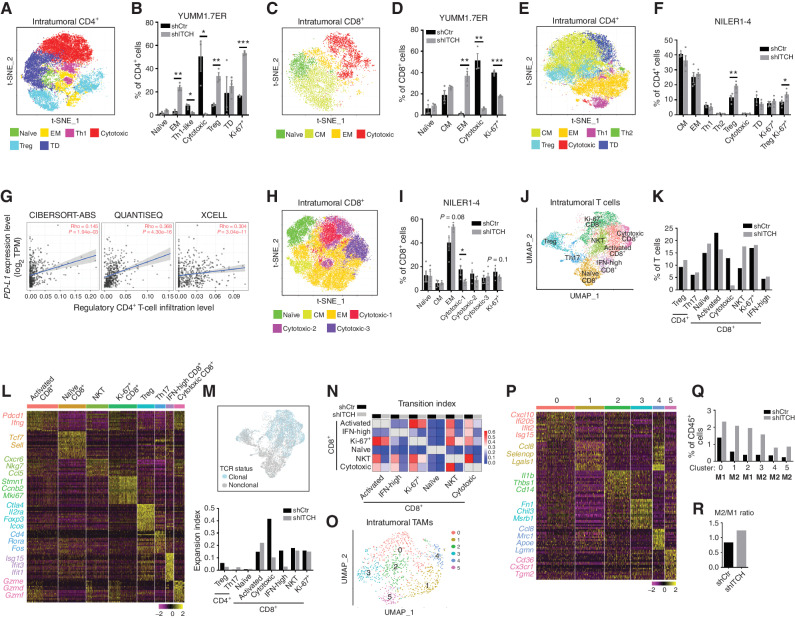
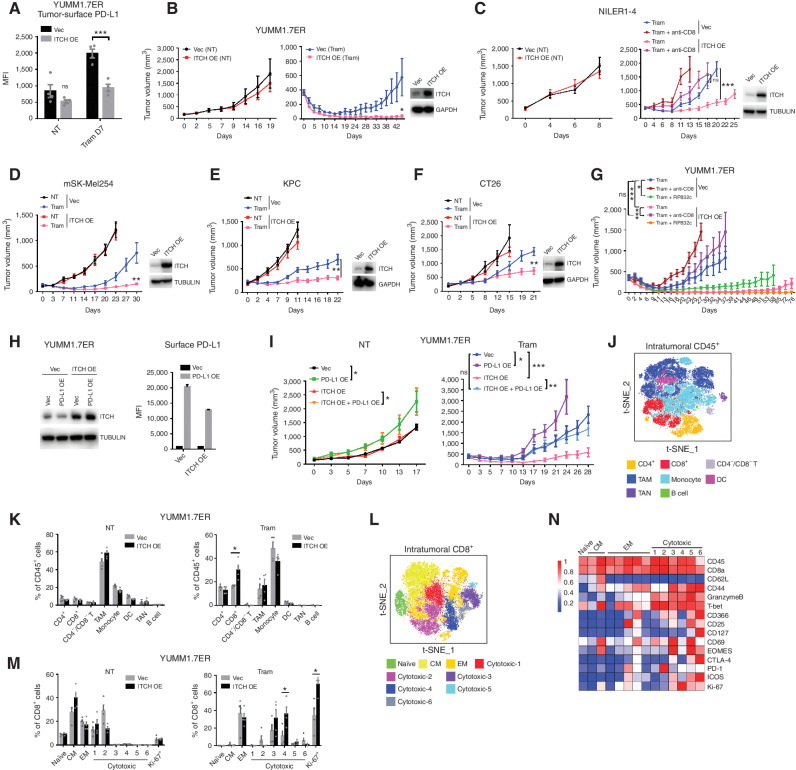
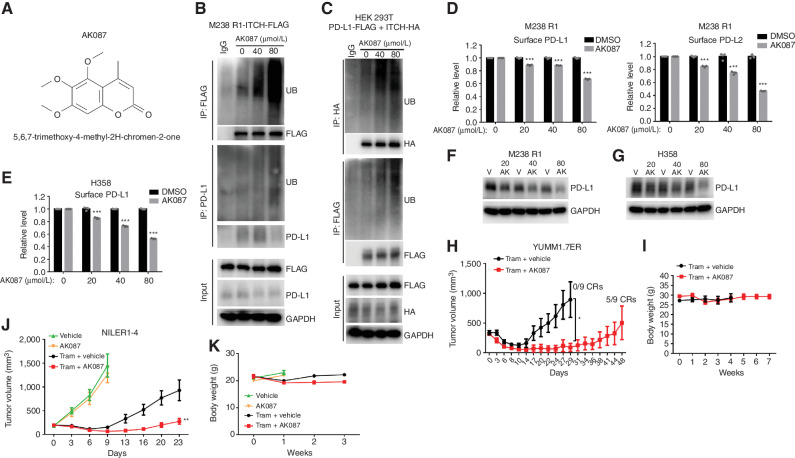
MAPK inhibitor (MAPKi) therapy in melanoma leads to the accumulation of tumor-surface PD-L1/L2, which may evade antitumor immunity and accelerate acquired resistance. Here, we discover that the E3 ligase ITCH binds, ubiquitinates, and downregulates tumor-surface PD-L1/L2 in MAPKi-treated human melanoma cells, thereby promoting T-cell activation. During MAPKi therapy in vivo, melanoma cell-intrinsic ITCH knockdown induced tumor-surface PD-L1, reduced intratumoral cytolytic CD8+ T cells, and accelerated acquired resistance only in immune-competent mice. Conversely, tumor cell-intrinsic ITCH overexpression reduced MAPKi-elicited PD-L1 accumulation, augmented intratumoral cytolytic CD8+ T cells, and suppressed acquired resistance in BrafV600MUT, NrasMUT, or Nf1MUT melanoma and KrasMUT-driven cancers. CD8+ T-cell depletion and tumor cell-intrinsic PD-L1 overexpression nullified the phenotype of ITCH overexpression, thereby supporting an in vivo ITCH-PD-L1-T-cell regulatory axis. Moreover, we identify a small-molecular ITCH activator that suppresses acquired MAPKi resistance in vivo. Thus, MAPKi-induced PD-L1 accelerates resistance, and a PD-L1-degrading ITCH activator prolongs antitumor response.
MAPKi induces tumor cell-surface PD-L1 accumulation, which promotes immune evasion and therapy resistance. ITCH degrades PD-L1, optimizing antitumor T-cell immunity. We propose degrading tumor cell-surface PD-L1 and/or activating tumor-intrinsic ITCH as strategies to overcome MAPKi resistance. This article is highlighted in the In This Issue feature, p. 1825.
在黑色素瘤中,MAPK 抑制剂(MAPKi)治疗会导致肿瘤表面 PD-L1/L2 的积累,这可能逃避抗肿瘤免疫并加速获得性耐药。在这里,我们发现 E3 连接酶 ITCH 结合、泛素化和下调 MAPKi 处理的人类黑色素瘤细胞中的肿瘤表面 PD-L1/L2,从而促进 T 细胞激活。在体内 MAPKi 治疗期间,黑色素瘤细胞内在的 ITCH 敲低诱导肿瘤表面 PD-L1,减少肿瘤内细胞毒性 CD8+T 细胞,并仅在免疫功能正常的小鼠中加速获得性耐药。相反,肿瘤细胞内在的 ITCH 过表达减少了 MAPKi 引发的 PD-L1 积累,增加了肿瘤内细胞毒性 CD8+T 细胞,并抑制了 BrafV600MUT、NrasMUT 或 Nf1MUT 黑色素瘤和 KrasMUT 驱动的癌症中的获得性耐药。CD8+T 细胞耗竭和肿瘤细胞内在的 PD-L1 过表达使 ITCH 过表达的表型无效,从而支持体内 ITCH-PD-L1-T 细胞调节轴。此外,我们鉴定了一种小分子 ITCH 激活剂,可在体内抑制获得性 MAPKi 耐药。因此,MAPKi 诱导的 PD-L1 加速耐药,而降解 PD-L1 的 ITCH 激活剂可延长抗肿瘤反应。
MAPKi 诱导肿瘤细胞表面 PD-L1 积累,促进免疫逃逸和治疗耐药。ITCH 降解 PD-L1,优化抗肿瘤 T 细胞免疫。我们提出降解肿瘤细胞表面 PD-L1 和/或激活肿瘤内在的 ITCH 作为克服 MAPKi 耐药的策略。本文在本期特色文章中重点介绍,第 1825 页。