Jo Ick-Hyun, Han Seahee, Shim Donghwan, Ryu Hojin, Hyun Tae Kyung, Lee Yi, Kim Daeil, So Yoon-Sup, Chung Jong-Wook
Department of Herbal Crop Research, National Institute of Horticultural and Herbal Science, Rural Development Administration, Eumseong, South Korea.
Division of Botany, Honam National Institute of Biological Resources, Mokpo, South Korea.
Front Plant Sci. 2022 May 16;13:891783. doi: 10.3389/fpls.2022.891783. eCollection 2022.
is an economically important forage crop in South Korea and China. Although detailed genetic and genomic data can improve population genetic studies, conservation efforts, and improved breeding of crops, few such data are available for species in general and none at all for . Therefore, the main objectives of this study were to sequence, assemble, and annotate chloroplast genome and to identify simple sequence repeats (SSRs) as polymorphic genetic markers.
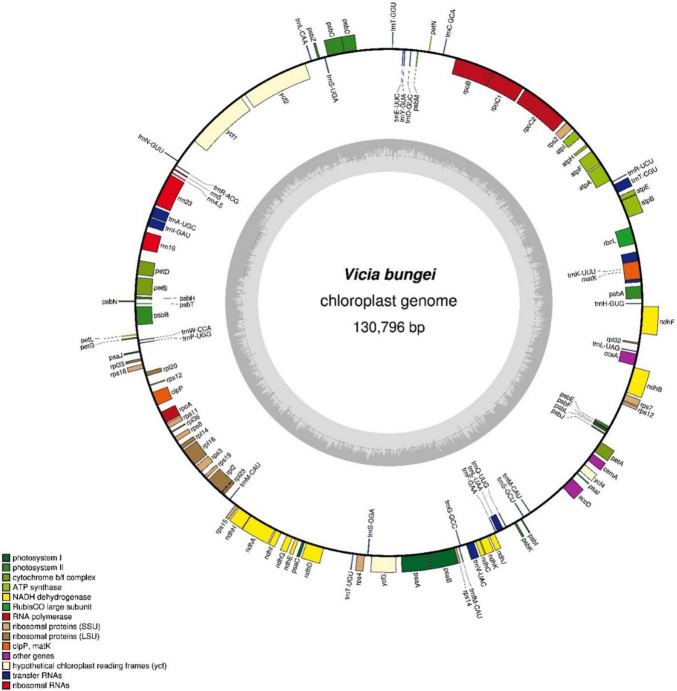
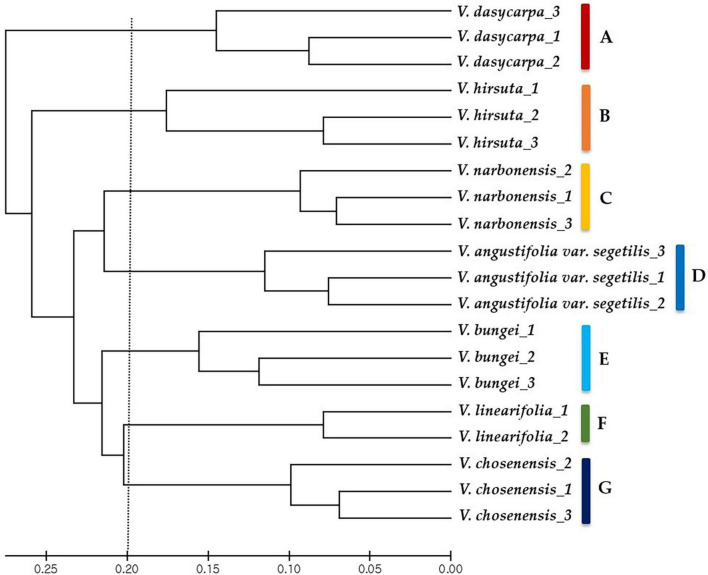
The whole-genome sequence of was generated using an Illumina MiSeq platform. De novo assembly of complete chloroplast genome sequences was performed for the low-coverage sequence using CLC Genome Assembler with a 200-600-bp overlap size. chloroplast genome was 130,796-bp long. The genome lacked an inverted repeat unit and thus resembled those of species in the inverted repeat-lacking clade within Fabaceae. Genome annotation using Dual OrganellarGenoMe Annotator (DOGMA) identified 107 genes, comprising 75 protein-coding, 28 transfer RNA, and 4 ribosomal RNA genes. In total, 432 SSRs were detected in chloroplast genome, including 64 mononucleotides, 14 dinucleotides, 5 trinucleotides, 4 tetranucleotides, 233 pentanucleotides, 90 hexanucleotides, and 14 complex repeated motifs. These were used to develop 232 novel chloroplast SSR markers, 39 of which were chosen at random to test amplification and genetic diversity in species (20 accessions from seven species). The unweighted pair group method with arithmetic mean cluster analysis identified seven clusters at the interspecies level and intraspecific differences within clusters.
The complete chloroplast genome sequence of was determined. This reference genome should facilitate chloroplast resequencing and future searches for additional genetic markers using population samples. The novel chloroplast genome resources and SSR markers will greatly contribute to the conservation of the genus and facilitate genetic and evolutionary studies of this genus and of other higher plants.
在韩国和中国是一种具有重要经济价值的饲料作物。尽管详细的遗传和基因组数据可以改善群体遗传学研究、保护工作以及作物的改良育种,但一般而言,关于该物种的此类数据很少,而关于[具体物种名称]的数据则完全没有。因此,本研究的主要目标是对[具体物种名称]叶绿体基因组进行测序、组装和注释,并鉴定简单序列重复(SSR)作为多态性遗传标记。
使用Illumina MiSeq平台生成了[具体物种名称]的全基因组序列。使用重叠大小为200 - 600 bp的CLC基因组组装器对低覆盖度序列进行了完整叶绿体基因组序列的从头组装。[具体物种名称]叶绿体基因组长度为130,796 bp。该基因组缺乏反向重复单元,因此类似于豆科植物中缺乏反向重复的分支中的物种。使用双细胞器基因组注释器(DOGMA)进行基因组注释,鉴定出107个基因,包括75个蛋白质编码基因、28个转运RNA基因和4个核糖体RNA基因。在[具体物种名称]叶绿体基因组中总共检测到432个SSR,包括64个单核苷酸、14个二核苷酸、5个三核苷酸、4个四核苷酸、233个五核苷酸、90个六核苷酸和14个复杂重复基序。这些被用于开发232个新的叶绿体SSR标记,随机选择其中39个来测试[具体物种名称]物种(来自七个物种的20个种质)中的扩增和遗传多样性。非加权组平均法聚类分析在种间水平鉴定出七个聚类以及聚类内的种内差异。
确定了[具体物种名称]的完整叶绿体基因组序列。该参考基因组应有助于叶绿体重测序以及未来使用群体样本寻找其他遗传标记。新的叶绿体基因组资源和SSR标记将极大地有助于[具体属名称]属的保护,并促进该属以及其他高等植物的遗传和进化研究。