Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA; Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
J Mol Biol. 2022 Jun 15;434(11):167530. doi: 10.1016/j.jmb.2022.167530. Epub 2022 Mar 5.
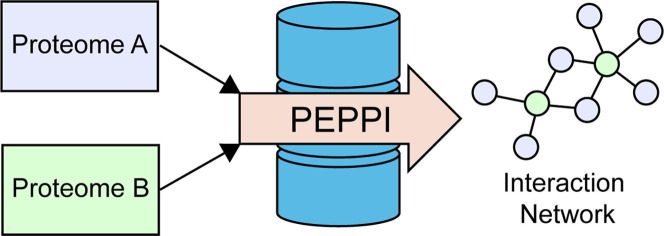
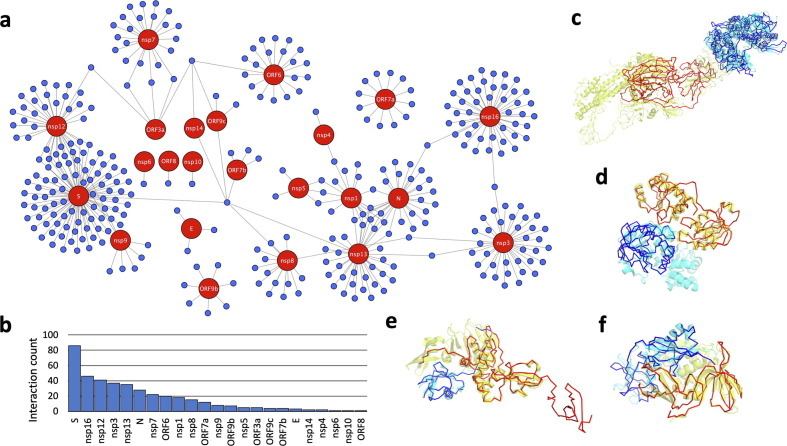
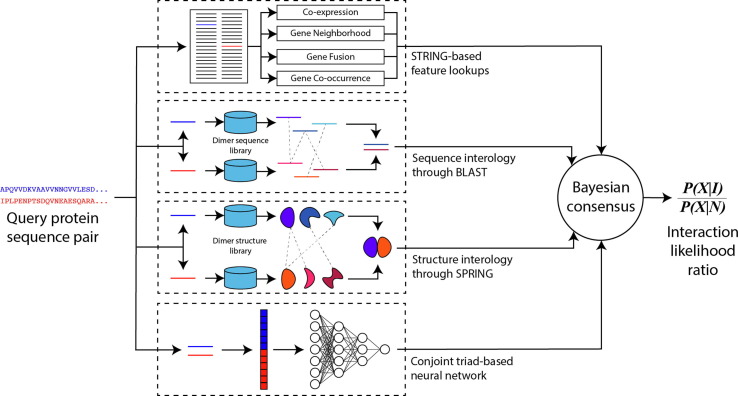
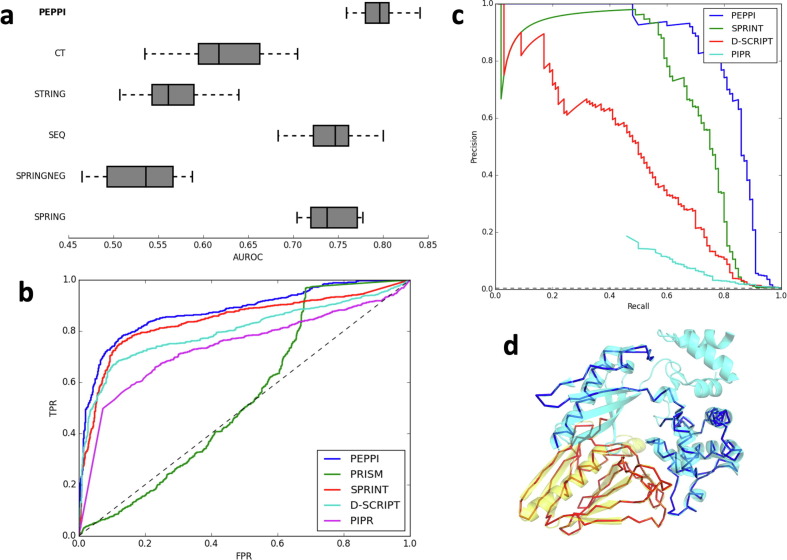
Proteome-wide identification of protein-protein interactions is a formidable task which has yet to be sufficiently addressed by experimental methodologies. Many computational methods have been developed to predict proteome-wide interaction networks, but few leverage both the sensitivity of structural information and the wide availability of sequence data. We present PEPPI, a pipeline which integrates structural similarity, sequence similarity, functional association data, and machine learning-based classification through a naïve Bayesian classifier model to accurately predict protein-protein interactions at a proteomic scale. Through benchmarking against a set of 798 ground truth interactions and an equal number of non-interactions, we have found that PEPPI attains 4.5% higher AUROC than the best of other state-of-the-art methods. As a proteomic-scale application, PEPPI was applied to model the interactions which occur between SARS-CoV-2 and human host cells during coronavirus infection, where 403 high-confidence interactions were identified with predictions covering 73% of a gold standard dataset from PSICQUIC and demonstrating significant complementarity with the most recent high-throughput experiments. PEPPI is available both as a webserver and in a standalone version and should be a powerful and generally applicable tool for computational screening of protein-protein interactions.
蛋白质组范围内的蛋白质-蛋白质相互作用的鉴定是一项艰巨的任务,目前还没有足够的实验方法来解决。已经开发了许多计算方法来预测蛋白质组范围内的相互作用网络,但很少有方法能够利用结构信息的敏感性和广泛可用的序列数据。我们提出了 PEPPI,这是一个集成了结构相似性、序列相似性、功能关联数据和基于机器学习的分类的管道,通过朴素贝叶斯分类器模型,可以在蛋白质组范围内准确预测蛋白质-蛋白质相互作用。通过与一组 798 个真实相互作用和数量相等的非相互作用进行基准测试,我们发现 PEPPI 的 AUROC 比其他最先进方法中的最佳方法高 4.5%。作为一种蛋白质组规模的应用,PEPPI 被应用于模拟 SARS-CoV-2 和人类宿主细胞在冠状病毒感染过程中发生的相互作用,其中鉴定了 403 个高可信度的相互作用,预测覆盖了 PSICQUIC 的黄金标准数据集的 73%,并与最近的高通量实验表现出显著的互补性。PEPPI 既提供了网络服务器版本,也提供了独立版本,应该是计算筛选蛋白质-蛋白质相互作用的强大而通用的工具。