School of Pharmacy, Dr. Vishwanath Karad MIT World Peace University, Pune, Maharashtra, 411038, India.
Bioinformatics Research Laboratory, Dr. D. Y. Patil Biotechnology & Bioinformatics Institute, Dr. D. Y. Patil Vidyapeeth, Pune, Maharashtra, 411033, India.
Comput Biol Med. 2022 Aug;147:105679. doi: 10.1016/j.compbiomed.2022.105679. Epub 2022 Jun 1.
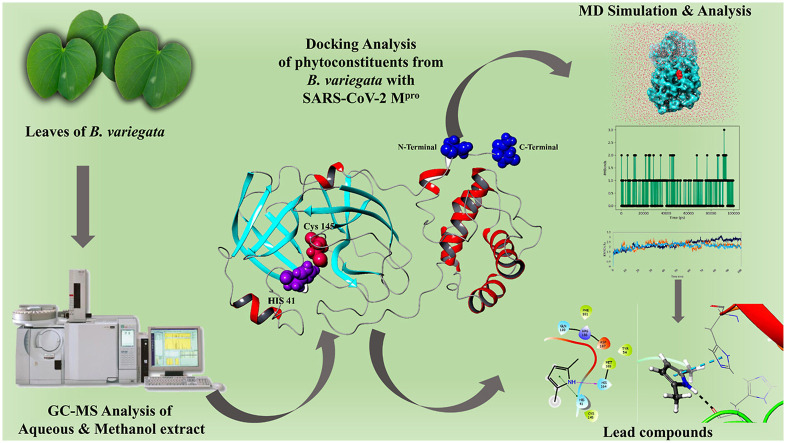
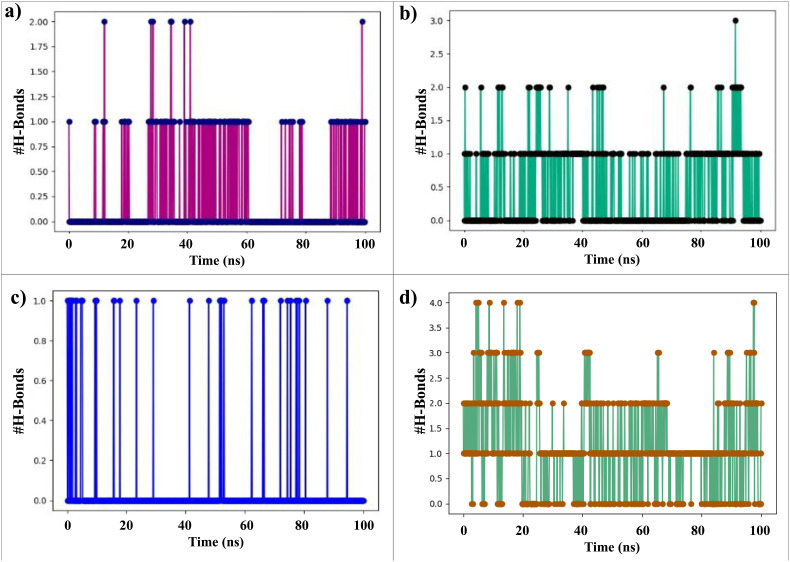
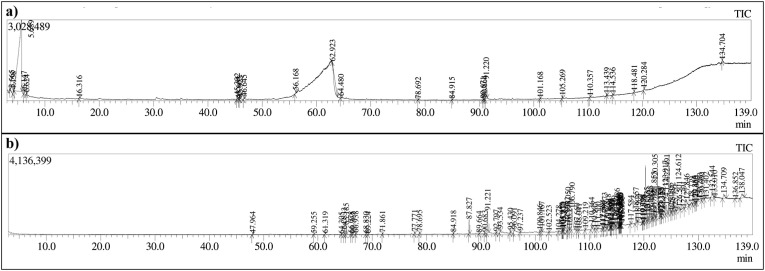
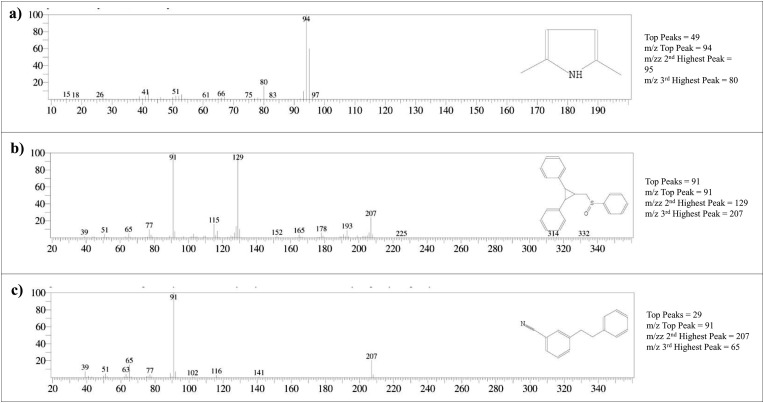
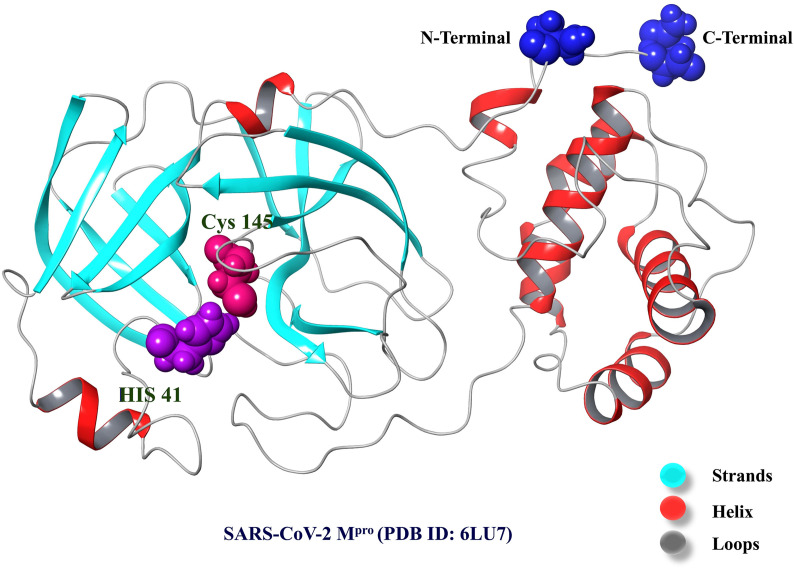
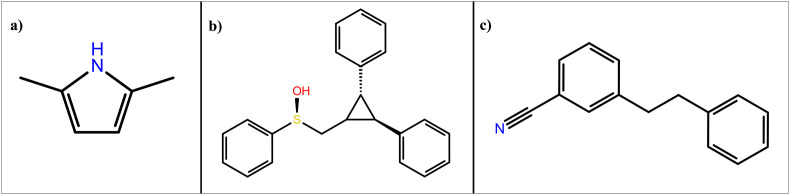
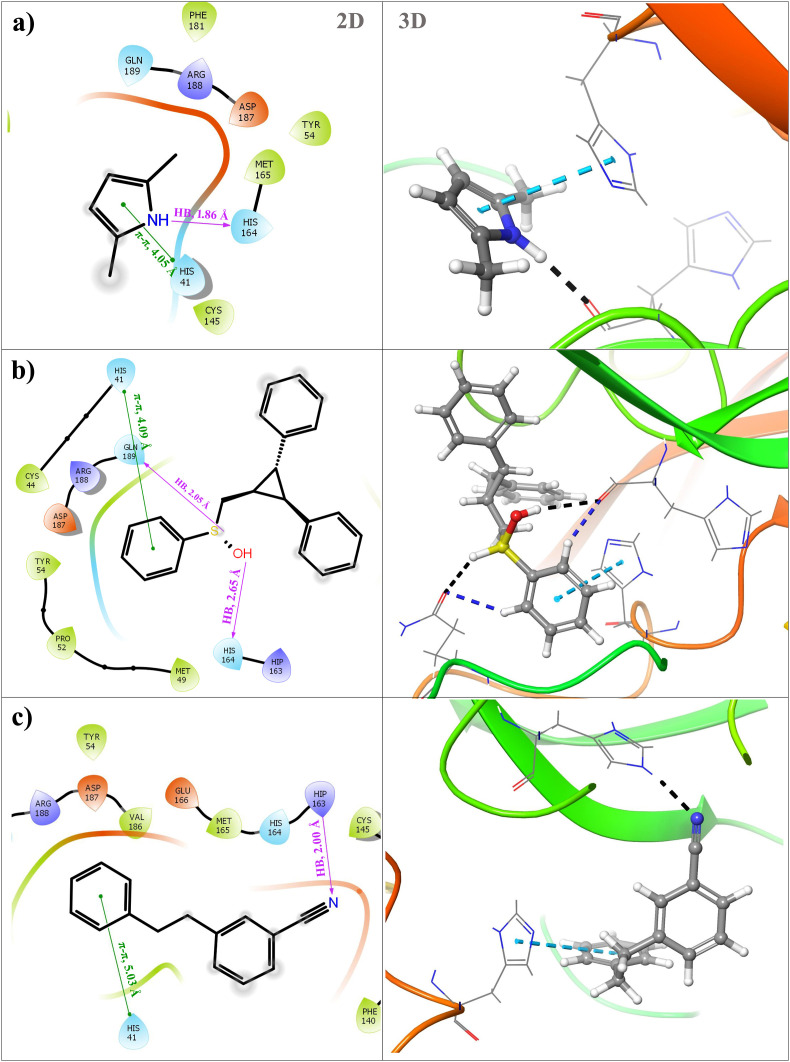
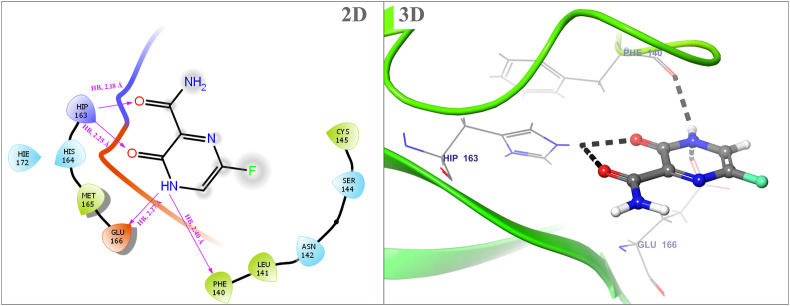
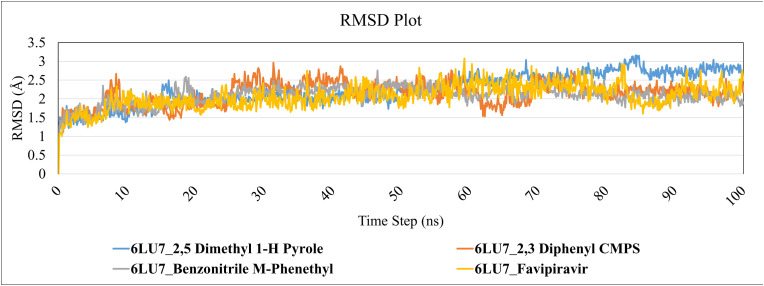
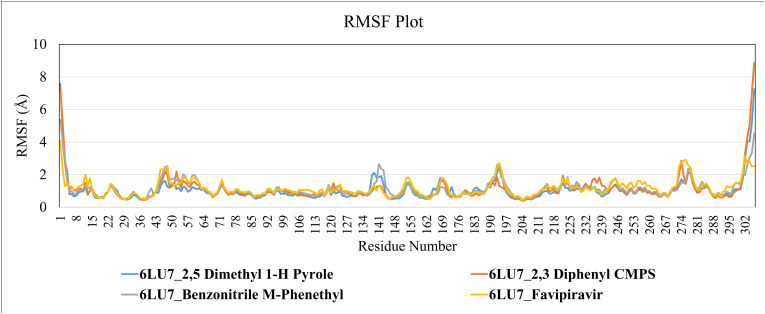
Severe acute respiratory syndrome coronavirus 2 was originally identified in Wuhan city of China in December 2019 and it spread rapidly throughout the globe, causing a threat to human life. Since targeted therapies are deficient, scientists all over the world have an opportunity to develop novel drug therapies to combat COVID-19. After the declaration of a global medical emergency, it was established that the Food and Drug Administration (FDA) could permit the use of emergency testing, treatments, and vaccines to decrease suffering, and loss of life, and restore the nation's health and security. The FDA has approved the use of remdesivir and its analogs as an antiviral medication, to treat COVID-19. The primary protease of SARS-CoV-2, which has the potential to regulate coronavirus proliferation, has been a viable target for the discovery of medicines against SARS-CoV-2. The present research deals with the in silico technique to screen phytocompounds from a traditional medicinal plant, Bauhinia variegata for potential inhibitors of the SARS-CoV-2 main protease. Dried leaves of the plant B. variegata were used to prepare aqueous and methanol extract and the constituents were analyzed using the GC-MS technique. A total of 57 compounds were retrieved from the aqueous and methanol extract analysis. Among these, three lead compounds (2,5 dimethyl 1-H Pyrrole, 2,3 diphenyl cyclopropyl methyl phenyl sulphoxide, and Benzonitrile m phenethyl) were shown to have the highest binding affinity (-5.719 to -5.580 kcal/mol) towards SARS-CoV-2 M. The post MD simulation results also revealed the favorable confirmation and stability of the selected lead compounds with M as per trajectory analysis. The Prime MM/GBSA binding free energy supports this finding, the top lead compound 2,3 diphenyl cyclopropyl methyl phenyl sulphoxide showed high binding free energy (-64.377 ± 5.24 kcal/mol) towards M which reflects the binding stability of the molecule with M. The binding free energy of the complexes was strongly influenced by His, Gln, and Glu residues. All of the molecules chosen are found to have strong pharmacokinetic characteristics and show drug-likeness properties. The lead compounds present acute toxicity (LD50) values ranging from 670 mg/kg to 2500 mg/kg; with toxicity classifications of 4 and 5 classes. Thus, these compounds could behave as probable lead candidates for treatment against SARS-CoV-2. However further in vitro and in vivo studies are required for the development of medication against SARS-CoV-2.
严重急性呼吸系统综合症冠状病毒 2 最初于 2019 年 12 月在中国武汉市被发现,它迅速在全球范围内传播,对人类生命构成威胁。由于缺乏靶向治疗方法,世界各地的科学家都有机会开发新的药物疗法来对抗 COVID-19。在宣布全球医疗紧急状态后,食品和药物管理局 (FDA) 可以允许使用紧急测试、治疗和疫苗来减少痛苦和生命损失,并恢复国家的健康和安全。FDA 已批准使用瑞德西韦及其类似物作为抗病毒药物,以治疗 COVID-19。SARS-CoV-2 的主要蛋白酶,具有调节冠状病毒增殖的潜力,一直是发现针对 SARS-CoV-2 的药物的可行目标。本研究采用计算机模拟技术,从一种传统药用植物白花紫荆中筛选出对 SARS-CoV-2 主要蛋白酶有潜在抑制作用的植物化合物。该植物的干叶被用来制备水提物和甲醇提取物,并使用 GC-MS 技术对其成分进行分析。从水提物和甲醇提取物分析中得到了 57 种化合物。其中,三种先导化合物(2,5 二甲基 1-H 吡咯、2,3 二苯基环丙基甲基苯基砜和苯甲腈间苯乙腈)对 SARS-CoV-2 M 表现出最高的结合亲和力(-5.719 至-5.580 kcal/mol)。MD 后模拟结果也表明,所选的先导化合物与 M 一起根据轨迹分析,具有有利的构象和稳定性。基于 Prime MM/GBSA 的结合自由能支持了这一发现,前导化合物 2,3 二苯基环丙基甲基苯基砜对 M 表现出高结合自由能(-64.377 ± 5.24 kcal/mol),反映了分子与 M 的结合稳定性。复合物的结合自由能受 His、Gln 和 Glu 残基的强烈影响。所选的所有分子都具有较强的药代动力学特性,并表现出类药性。先导化合物的急性毒性(LD50)值范围为 670mg/kg 至 2500mg/kg;毒性分类为 4 类和 5 类。因此,这些化合物可能成为治疗 SARS-CoV-2 的潜在候选药物。然而,还需要进一步的体外和体内研究来开发针对 SARS-CoV-2 的药物。