Deng Lei, Zeng Yunyun, Liu Hui, Liu Zixuan, Liu Xuejun
School of Computer Science and Engineering, Central South University, Changsha 410083, China.
School of Computer Science and Technology, Nanjing Tech University, Nanjing 211816, China.
Curr Issues Mol Biol. 2022 May 19;44(5):2287-2299. doi: 10.3390/cimb44050155.
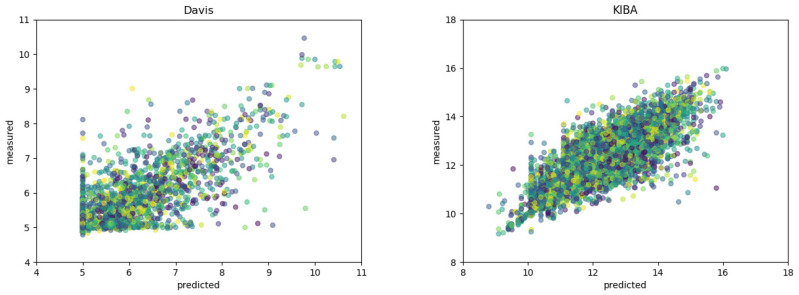
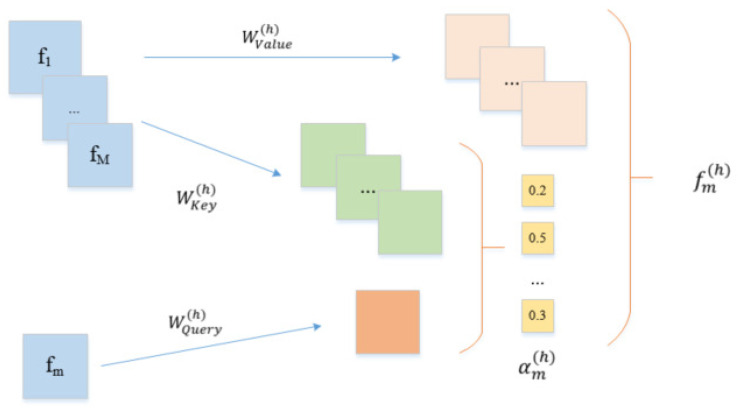
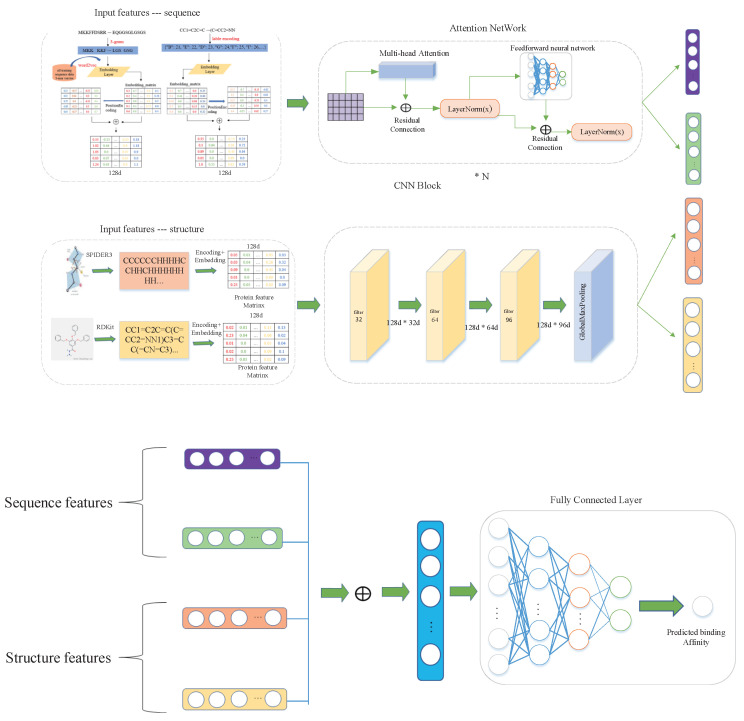
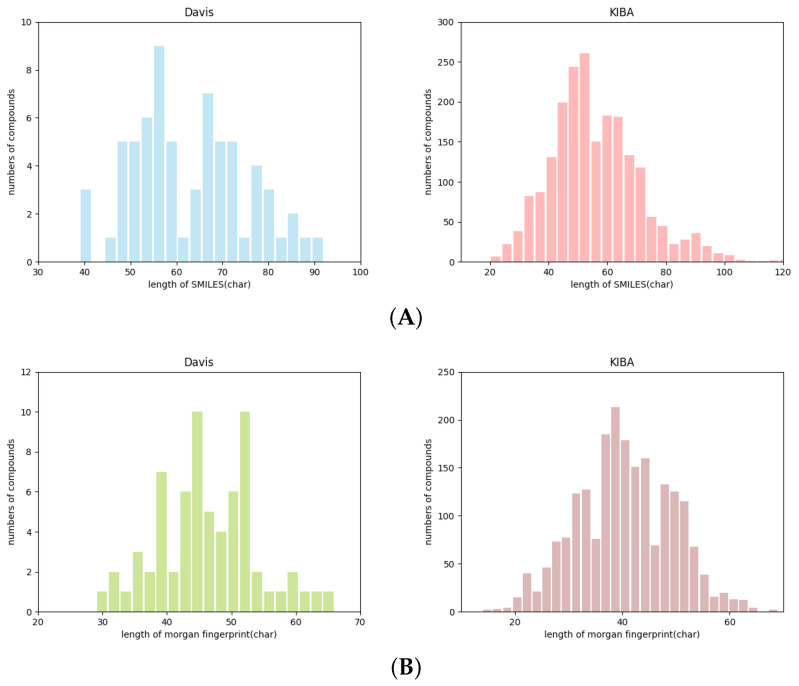
Drug-target interactions provide insight into the drug-side effects and drug repositioning. However, wet-lab biochemical experiments are time-consuming and labor-intensive, and are insufficient to meet the pressing demand for drug research and development. With the rapid advancement of deep learning, computational methods are increasingly applied to screen drug-target interactions. Many methods consider this problem as a binary classification task (binding or not), but ignore the quantitative binding affinity. In this paper, we propose a new end-to-end deep learning method called DeepMHADTA, which uses the multi-head self-attention mechanism in a deep residual network to predict drug-target binding affinity. On two benchmark datasets, our method outperformed several current state-of-the-art methods in terms of multiple performance measures, including mean square error (MSE), consistency index (CI), rm2, and PR curve area (AUPR). The results demonstrated that our method achieved better performance in predicting the drug-target binding affinity.
药物-靶点相互作用有助于深入了解药物副作用和药物重新定位。然而,湿实验室生化实验既耗时又费力,且不足以满足药物研发的迫切需求。随着深度学习的快速发展,计算方法越来越多地应用于筛选药物-靶点相互作用。许多方法将此问题视为二元分类任务(结合或不结合),但忽略了定量结合亲和力。在本文中,我们提出了一种名为DeepMHADTA的新型端到端深度学习方法,该方法在深度残差网络中使用多头自注意力机制来预测药物-靶点结合亲和力。在两个基准数据集上,我们的方法在包括均方误差(MSE)、一致性指数(CI)、rm2和PR曲线面积(AUPR)在内的多个性能指标方面优于当前几种最先进的方法。结果表明,我们的方法在预测药物-靶点结合亲和力方面取得了更好的性能。