Department of Biochemistry and Biophysics, National Bioinformatics Infrastructure Sweden, Science for Life Laboratory, Stockholm University, Solna, Sweden.
Department of Cell and Molecular Biology, National Bioinformatics Infrastructure Sweden, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
BMC Bioinformatics. 2022 Jun 13;23(1):228. doi: 10.1186/s12859-022-04757-0.
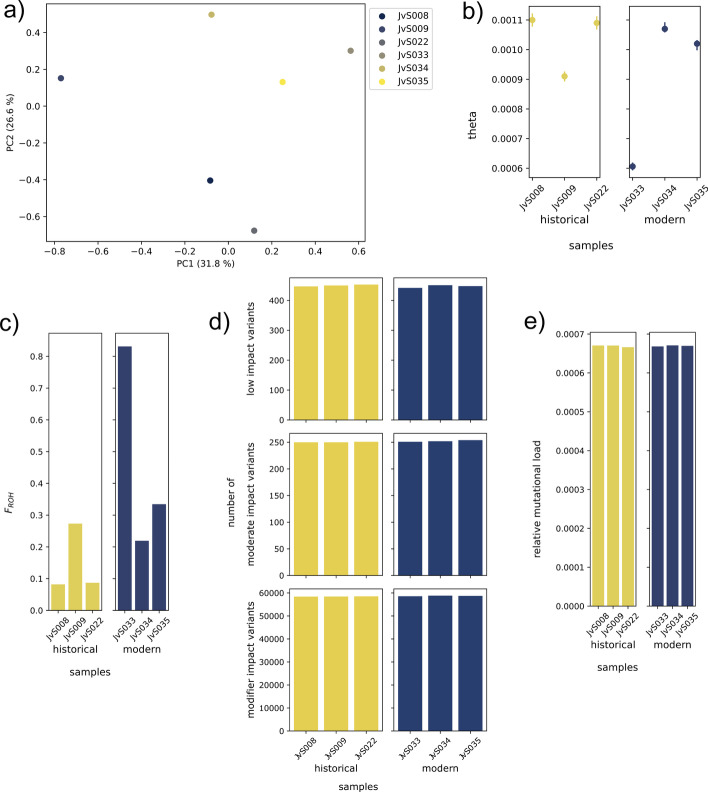
Many wild species have suffered drastic population size declines over the past centuries, which have led to 'genomic erosion' processes characterized by reduced genetic diversity, increased inbreeding, and accumulation of harmful mutations. Yet, genomic erosion estimates of modern-day populations often lack concordance with dwindling population sizes and conservation status of threatened species. One way to directly quantify the genomic consequences of population declines is to compare genome-wide data from pre-decline museum samples and modern samples. However, doing so requires computational data processing and analysis tools specifically adapted to comparative analyses of degraded, ancient or historical, DNA data with modern DNA data as well as personnel trained to perform such analyses.
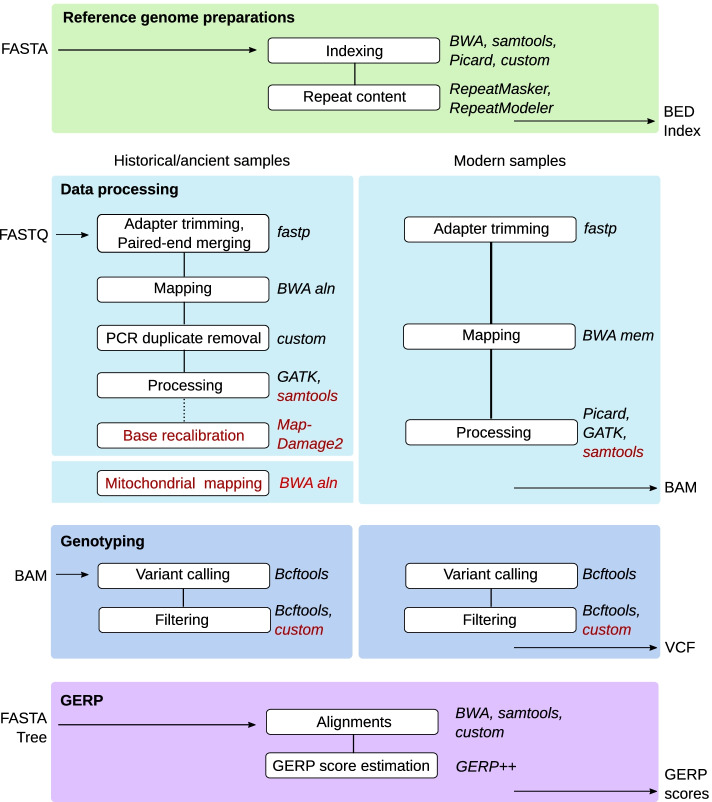
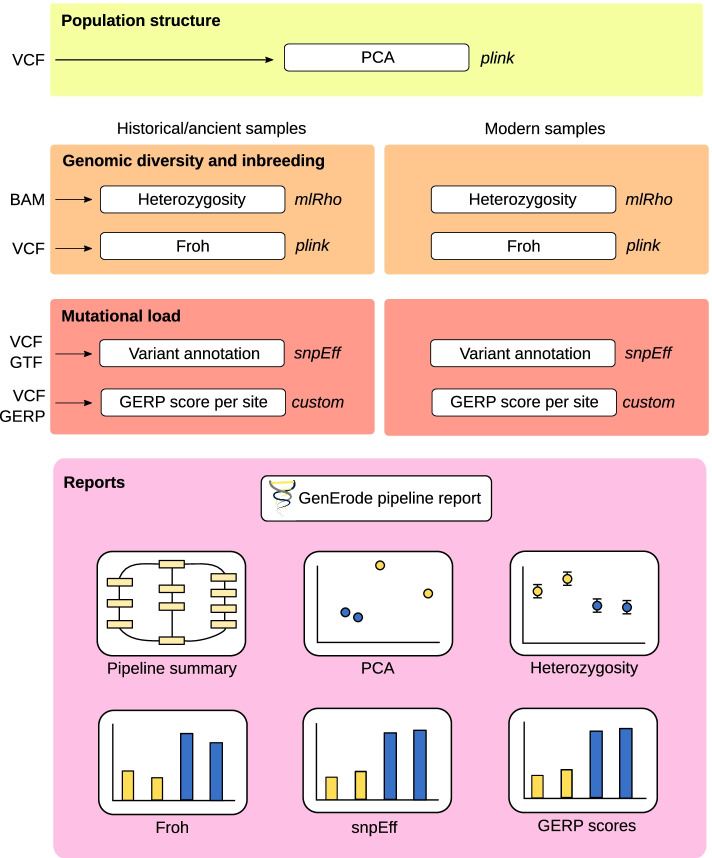
Here, we present a highly flexible, scalable, and modular pipeline to compare patterns of genomic erosion using samples from disparate time periods. The GenErode pipeline uses state-of-the-art bioinformatics tools to simultaneously process whole-genome re-sequencing data from ancient/historical and modern samples, and to produce comparable estimates of several genomic erosion indices. No programming knowledge is required to run the pipeline and all bioinformatic steps are well-documented, making the pipeline accessible to users with different backgrounds. GenErode is written in Snakemake and Python3 and uses Conda and Singularity containers to achieve reproducibility on high-performance compute clusters. The source code is freely available on GitHub ( https://github.com/NBISweden/GenErode ).
GenErode is a user-friendly and reproducible pipeline that enables the standardization of genomic erosion indices from temporally sampled whole genome re-sequencing data.
在过去的几个世纪里,许多野生物种的种群数量急剧减少,导致了“基因组侵蚀”过程,其特征是遗传多样性减少、近亲繁殖增加和有害突变积累。然而,现代种群的基因组侵蚀估计往往与濒危物种的种群减少和保护状况缺乏一致性。一种直接量化种群减少对基因组影响的方法是比较衰退前博物馆样本和现代样本的全基因组数据。然而,这样做需要专门用于比较降解、古老或历史 DNA 数据与现代 DNA 数据的计算数据处理和分析工具,以及受过执行此类分析培训的人员。
在这里,我们提出了一个高度灵活、可扩展和模块化的管道,用于使用来自不同时期的样本比较基因组侵蚀模式。GenErode 管道使用最先进的生物信息学工具,同时处理来自古代/历史和现代样本的全基因组重测序数据,并生成几个基因组侵蚀指数的可比估计值。运行管道不需要编程知识,并且所有生物信息学步骤都有详细的文档记录,这使得不同背景的用户都可以使用该管道。GenErode 是用 Snakemake 和 Python3 编写的,并使用 Conda 和 Singularity 容器在高性能计算集群上实现可重复性。源代码可在 GitHub 上免费获得(https://github.com/NBISweden/GenErode)。
GenErode 是一个用户友好且可重复的管道,它能够标准化来自时间采样的全基因组重测序数据的基因组侵蚀指数。