J. Craig Venter Institute, La Jolla, CA, USA.
Chan Zuckerberg Initiative, Redwood City, CA, USA.
Sci Rep. 2022 Jun 15;12(1):9996. doi: 10.1038/s41598-022-14192-z.
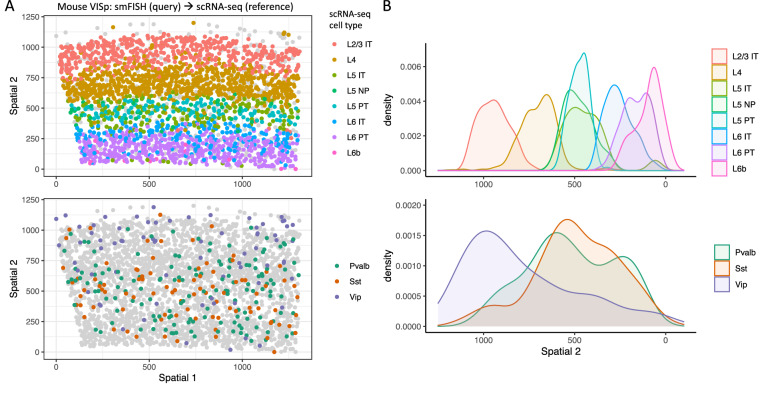
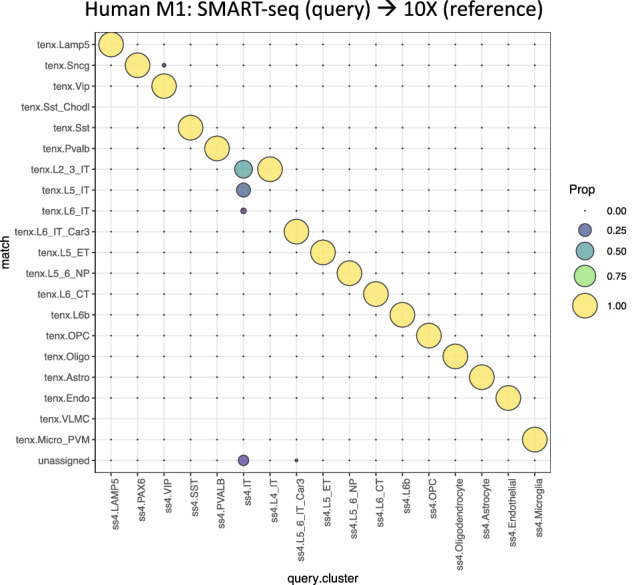
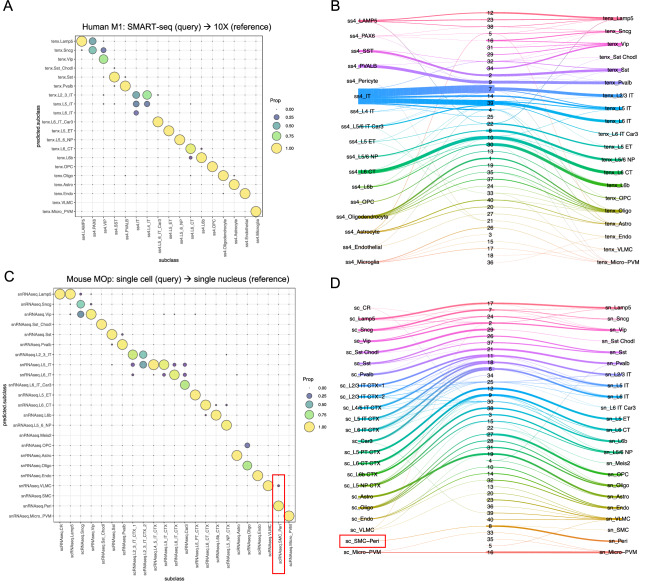
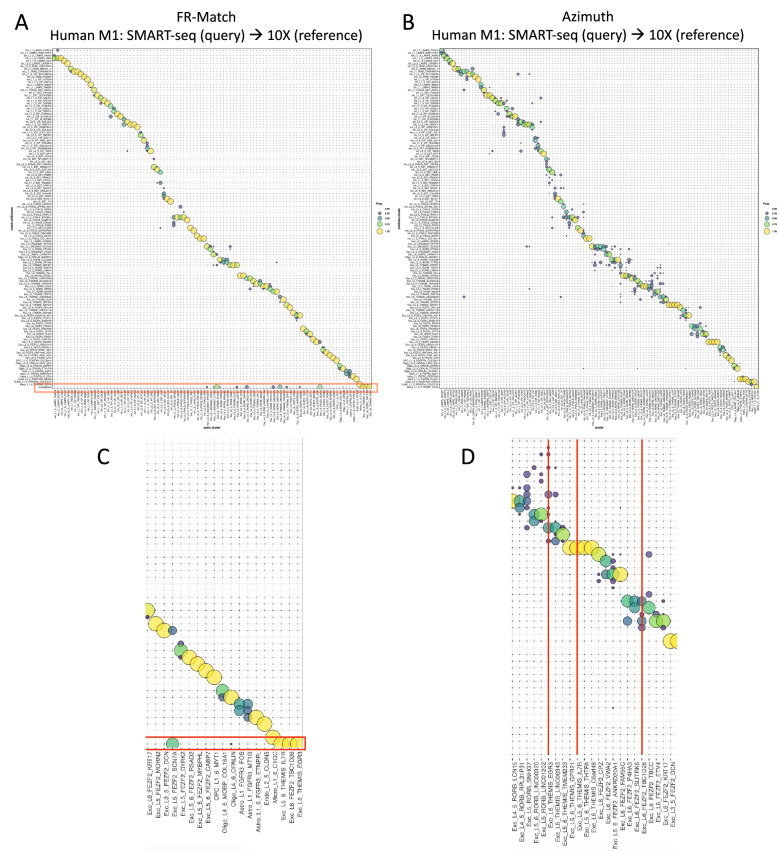
Reference cell atlases powered by single cell and spatial transcriptomics technologies are becoming available to study healthy and diseased tissue at single cell resolution. One important use of these data resources is to compare cell types from new dataset with cell types in the reference atlases to evaluate their phenotypic similarities and differences, for example, for identifying novel cell types under disease conditions. For this purpose, rigorously-validated computational algorithms are needed to perform these cell type matching tasks that can compare datasets from different experiment platforms and sample types. Here, we present significant enhancements to FR-Match (v2.0)-a multivariate nonparametric statistical testing approach for matching cell types in query datasets to reference atlases. FR-Match v2.0 includes a normalization procedure to facilitate cross-platform cluster-level comparisons (e.g., plate-based SMART-seq and droplet-based 10X Chromium single cell and single nucleus RNA-seq and spatial transcriptomics) and extends the pipeline to also allow cell-level matching. In the use cases evaluated, FR-Match showed robust and accurate performance for identifying common and novel cell types across tissue regions, for discovering sub-optimally clustered cell types, and for cross-platform and cross-sample cell type matching.
基于单细胞和空间转录组学技术的参考细胞图谱可用于以单细胞分辨率研究健康和患病组织。这些数据资源的一个重要用途是将新数据集的细胞类型与参考图谱中的细胞类型进行比较,以评估它们的表型相似性和差异,例如,用于在疾病条件下识别新的细胞类型。为此,需要经过严格验证的计算算法来执行这些细胞类型匹配任务,以比较来自不同实验平台和样本类型的数据集。在这里,我们对 FR-Match(v2.0)进行了重大改进 - 这是一种用于将查询数据集中的细胞类型与参考图谱进行匹配的多变量非参数统计测试方法。FR-Match v2.0 包括一个标准化程序,以促进跨平台的聚类级比较(例如,基于平板的 SMART-seq 和基于液滴的 10X Chromium 单细胞和单核 RNA-seq 和空间转录组学),并扩展了该管道以允许进行细胞级匹配。在评估的用例中,FR-Match 在识别跨组织区域的常见和新型细胞类型、发现聚类效果不佳的细胞类型以及跨平台和跨样本的细胞类型匹配方面表现出稳健和准确的性能。