College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China.
BGI Research, Hangzhou 310012, China.
Brief Bioinform. 2024 May 23;25(4). doi: 10.1093/bib/bbae250.
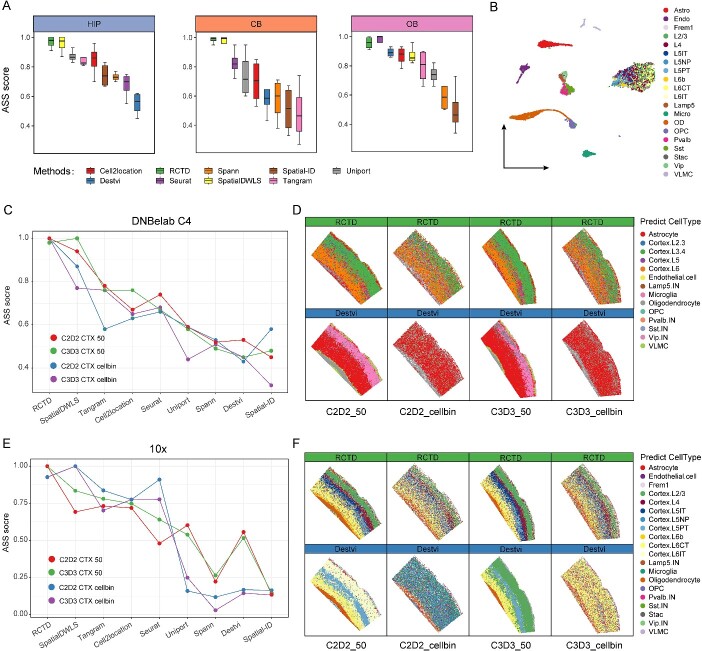
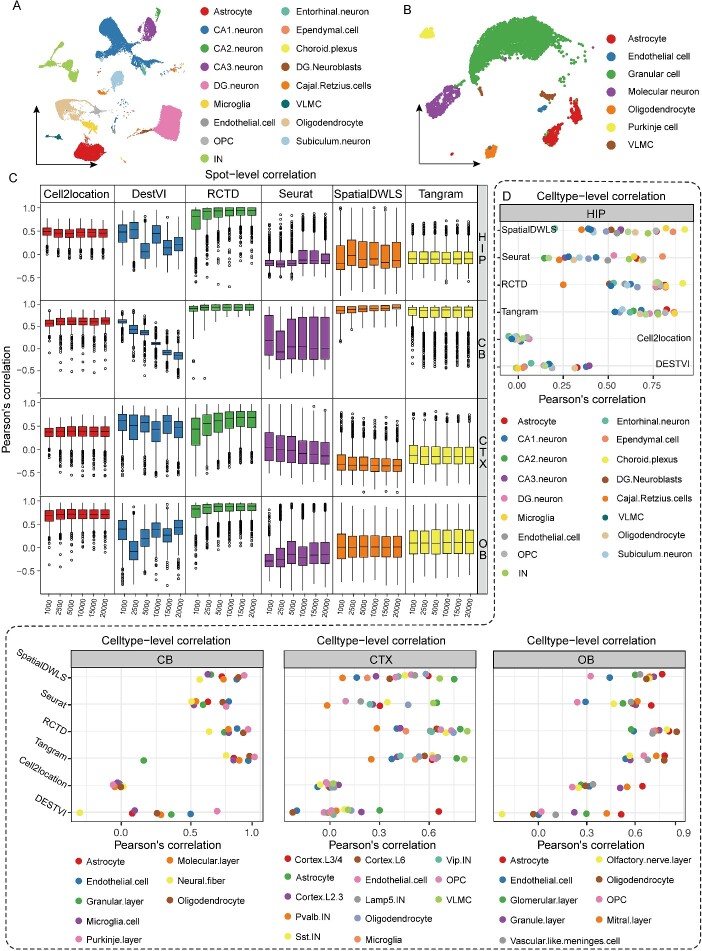
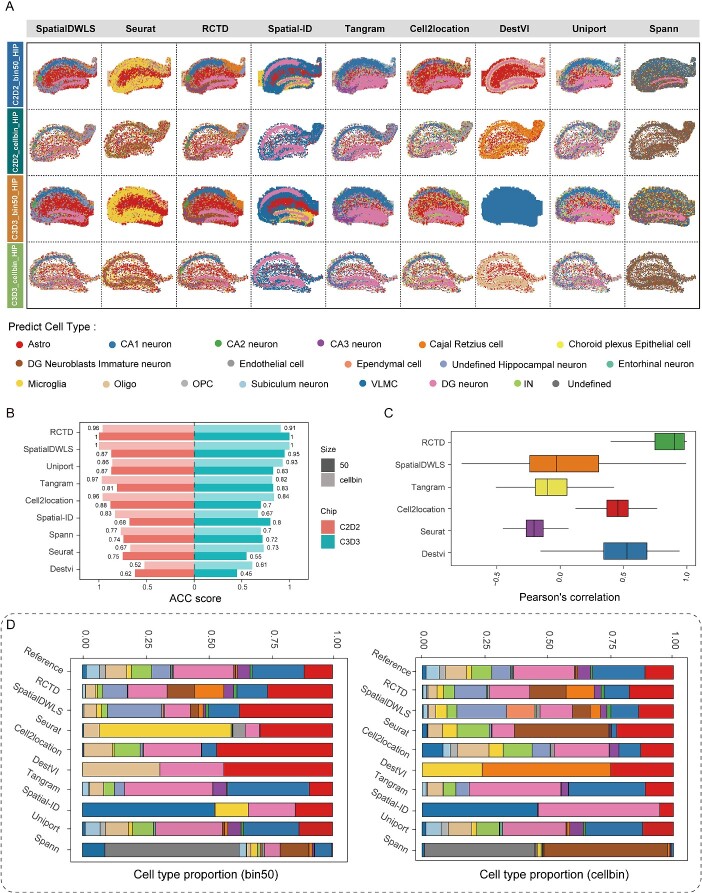
Limited gene capture efficiency and spot size of spatial transcriptome (ST) data pose significant challenges in cell-type characterization. The heterogeneity and complexity of cell composition in the mammalian brain make it more challenging to accurately annotate ST data from brain. Many algorithms attempt to characterize subtypes of neuron by integrating ST data with single-nucleus RNA sequencing (snRNA-seq) or single-cell RNA sequencing. However, assessing the accuracy of these algorithms on Stereo-seq ST data remains unresolved. Here, we benchmarked 9 mapping algorithms using 10 ST datasets from four mouse brain regions in two different resolutions and 24 pseudo-ST datasets from snRNA-seq. Both actual ST data and pseudo-ST data were mapped using snRNA-seq datasets from the corresponding brain regions as reference data. After comparing the performance across different areas and resolutions of the mouse brain, we have reached the conclusion that both robust cell-type decomposition and SpatialDWLS demonstrated superior robustness and accuracy in cell-type annotation. Testing with publicly available snRNA-seq data from another sequencing platform in the cortex region further validated our conclusions. Altogether, we developed a workflow for assessing suitability of mapping algorithm that fits for ST datasets, which can improve the efficiency and accuracy of spatial data annotation.
空间转录组(ST)数据的基因捕获效率有限且斑点大小有限,这对细胞类型特征描述构成了重大挑战。哺乳动物大脑中细胞组成的异质性和复杂性使得更难以准确注释来自大脑的 ST 数据。许多算法试图通过将 ST 数据与单核 RNA 测序(snRNA-seq)或单细胞 RNA 测序相结合来描述神经元亚型。然而,评估这些算法在 Stereo-seq ST 数据上的准确性仍然没有得到解决。在这里,我们使用来自两个不同分辨率的四个小鼠大脑区域的 10 个 ST 数据集和来自 snRNA-seq 的 24 个伪 ST 数据集,对 9 种映射算法进行了基准测试。使用来自相应大脑区域的 snRNA-seq 数据集作为参考数据,对实际的 ST 数据和伪 ST 数据进行了映射。在比较了不同区域和分辨率的小鼠大脑的性能之后,我们得出结论,稳健的细胞类型分解和 SpatialDWLS 在细胞类型注释方面都表现出了更好的稳健性和准确性。在皮质区域使用另一个测序平台提供的公开可用的 snRNA-seq 数据进行测试进一步验证了我们的结论。总的来说,我们开发了一种评估适合 ST 数据集的映射算法适用性的工作流程,这可以提高空间数据注释的效率和准确性。