Amdur M Jeremy, Mullin Kathleen R, Waters Michael J, Puggioni Danilo, Wojnar Michael K, Gu Mingqiang, Sun Lei, Oyala Paul H, Rondinelli James M, Freedman Danna E
Department of Chemistry, Massachusetts Institute of Technology Cambridge Massachusetts 02139 USA
Department of Materials Science and Engineering, Northwestern University Evanston Illinois 60208 USA
Chem Sci. 2022 May 17;13(23):7034-7045. doi: 10.1039/d1sc06130e. eCollection 2022 Jun 15.
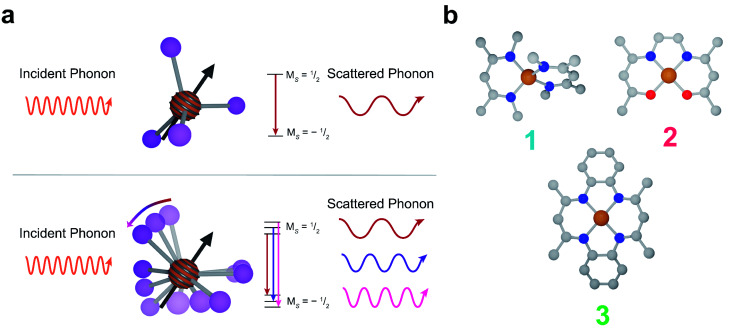
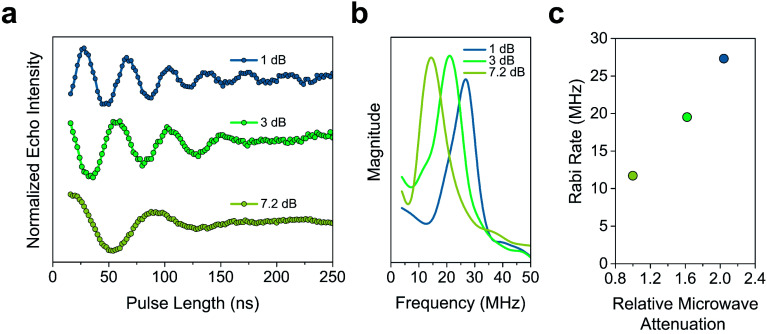
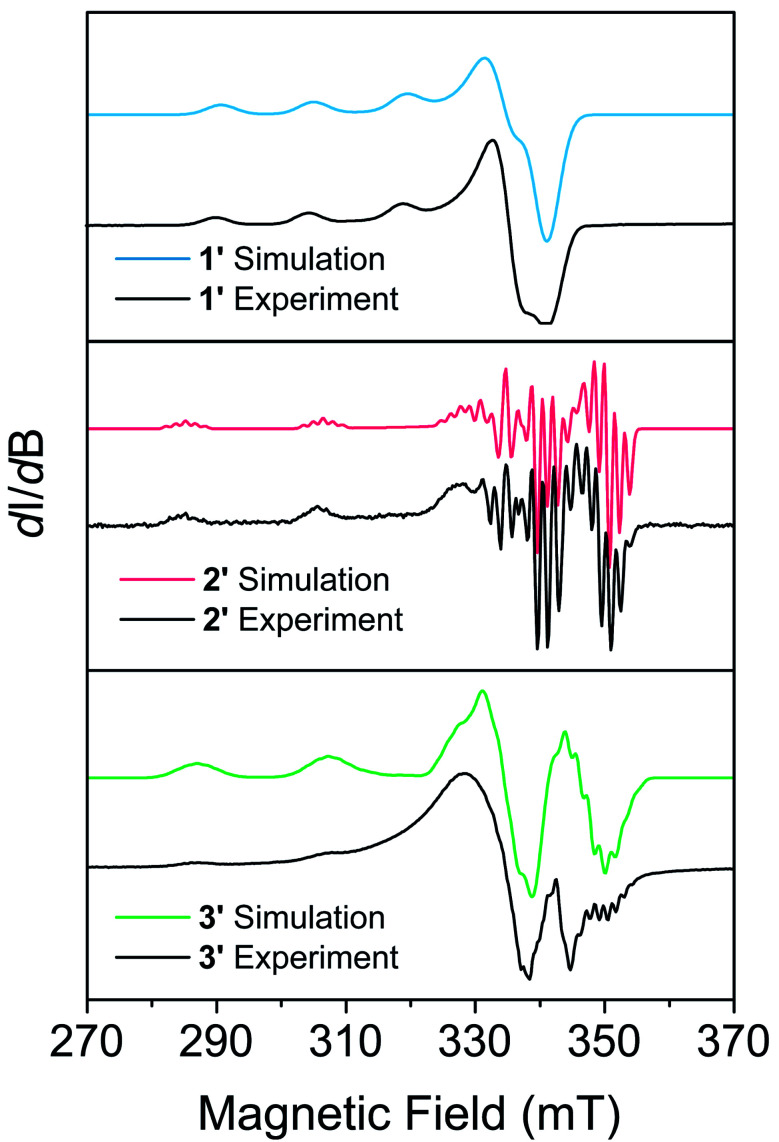
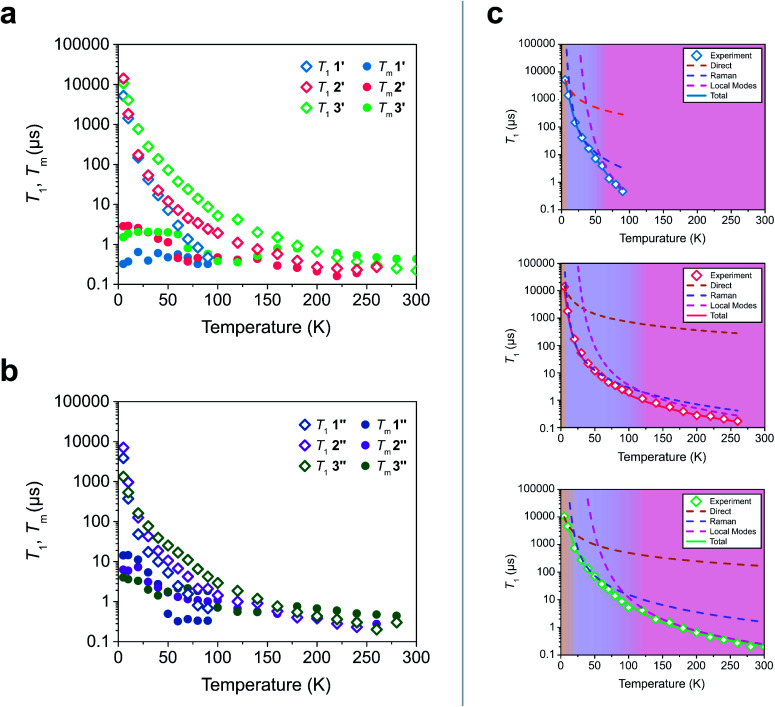
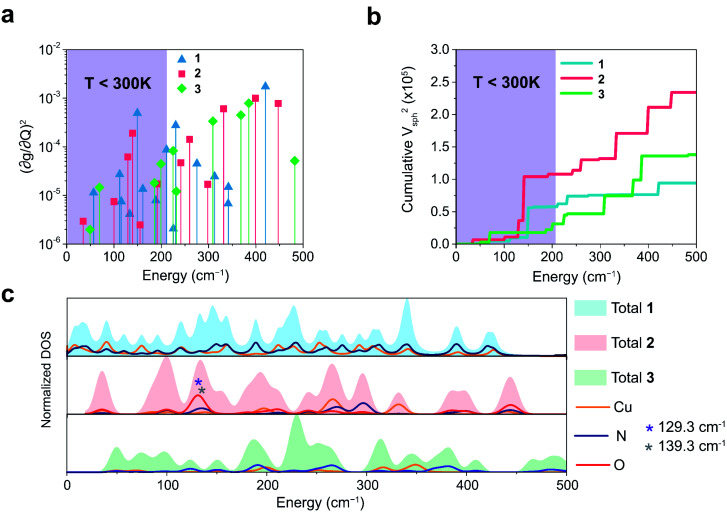
The second quantum revolution harnesses exquisite quantum control for a slate of diverse applications including sensing, communication, and computation. Of the many candidates for building quantum systems, molecules offer both tunability and specificity, but the principles to enable high temperature operation are not well established. Spin-lattice relaxation, represented by the time constant , is the primary factor dictating the high temperature performance of quantum bits (qubits), and serves as the upper limit on qubit coherence times ( ). For molecular qubits at elevated temperatures (>100 K), molecular vibrations facilitate rapid spin-lattice relaxation which limits to well below operational minimums for certain quantum technologies. Here we identify the effects of controlling orbital angular momentum through metal coordination geometry and ligand rigidity π-conjugation on relaxation in three four-coordinate Cu = ½ qubit candidates: bis(,'-dimethyl-4-amino-3-penten-2-imine) copper(ii) (MeNac) (1), bis(acetylacetone)ethylenediamine copper(ii) Cu(acacen) (2), and tetramethyltetraazaannulene copper(ii) Cu(tmtaa) (3). We obtain significant improvement upon changing from tetrahedral to square planar geometries through changes in orbital angular momentum. is further improved with greater π-conjugation in the ligand framework. Our electronic structure calculations reveal that the reduced motion of low energy vibrations in the primary coordination sphere slows relaxation and increases . These principles enable us to report a new molecular qubit candidate with room temperature = 0.43 μs, and establishes guidelines for designing novel qubit candidates operating above 100 K.
第二次量子革命利用精确的量子控制实现了一系列不同的应用,包括传感、通信和计算。在构建量子系统的众多候选材料中,分子兼具可调性和特异性,但实现高温运行的原理尚未完全确立。以时间常数 表示的自旋 - 晶格弛豫是决定量子比特(qubit)高温性能的主要因素,也是量子比特相干时间( )的上限。对于高温(>100 K)下的分子量子比特,分子振动会促进快速的自旋 - 晶格弛豫,这将 限制在远低于某些量子技术运行最小值的水平。在此,我们确定了通过金属配位几何结构和配体刚性π共轭来控制轨道角动量对三种四配位Cu(自旋 = ½)量子比特候选物中 弛豫的影响:双(,' - 二甲基 - 4 - 氨基 - 3 - 戊烯 - 2 - 亚胺)铜(ii)(MeNac)(1)、双(乙酰丙酮)乙二胺铜(ii)Cu(acacen)(2)和四甲基四氮杂环壬四烯铜(ii)Cu(tmtaa)(3)。通过改变轨道角动量,从四面体几何结构转变为平面正方形几何结构时,我们实现了 的显著改善。配体框架中更大的π共轭进一步改善了 。我们的电子结构计算表明,初级配位球中低能振动的运动减少会减缓弛豫并增加 。这些原理使我们能够报道一种新的分子量子比特候选物,其室温下的 = 0.43 μs,并为设计在100 K以上运行的新型量子比特候选物建立了指导原则。