Institut Pasteur, Université Paris Cité, Unité Bioinformatique Evolutive, Paris, France.
Université de Paris, Paris, France.
Nat Commun. 2022 Jul 6;13(1):3896. doi: 10.1038/s41467-022-31511-0.
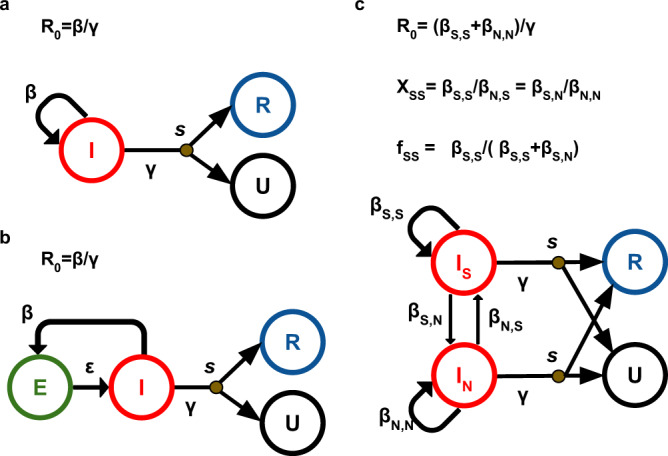
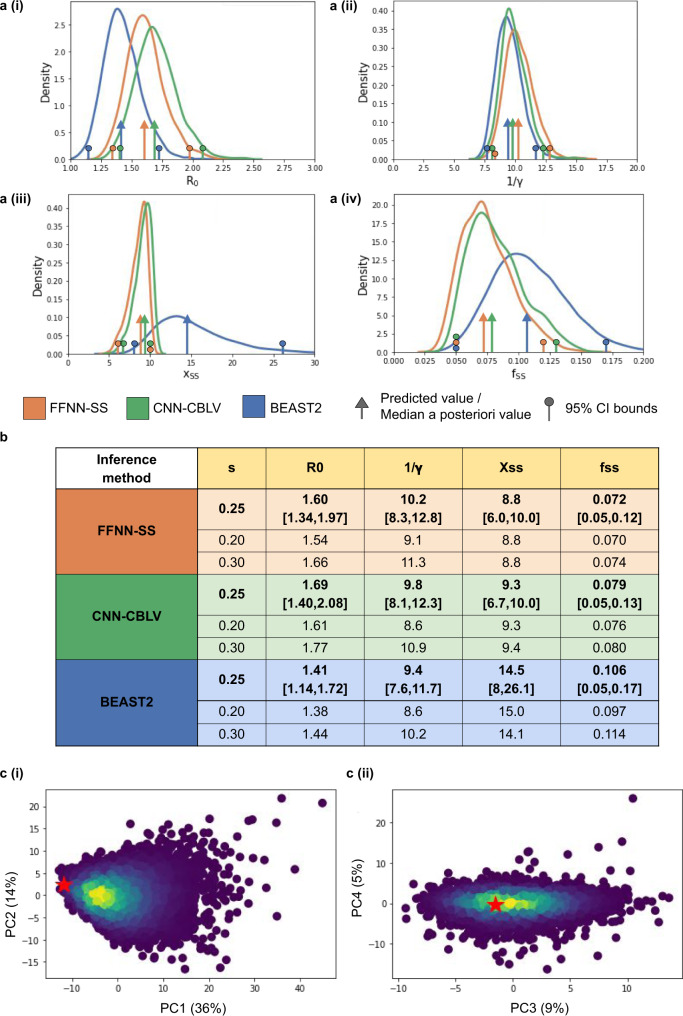
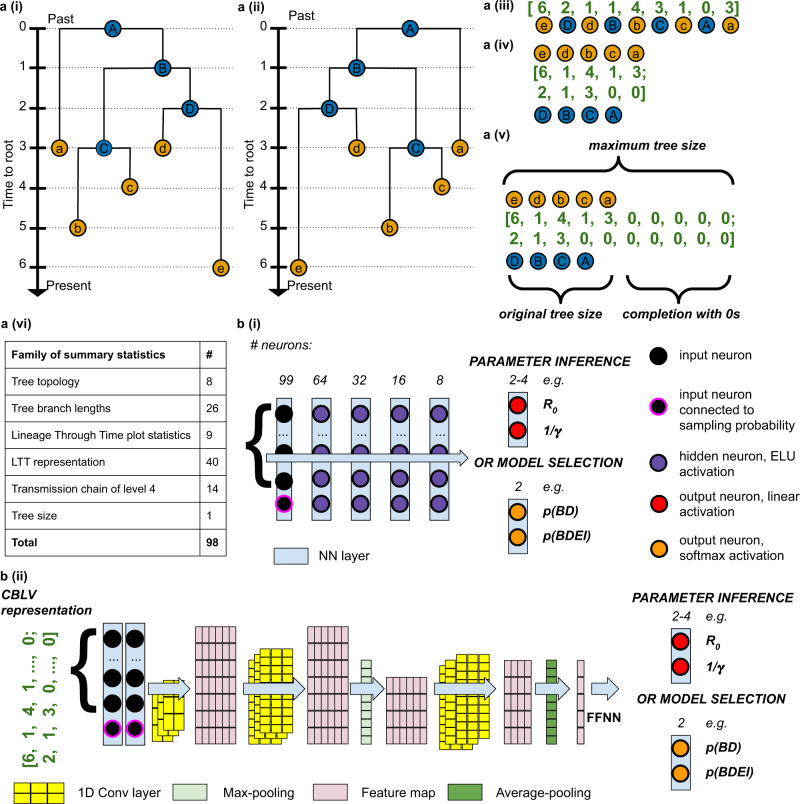
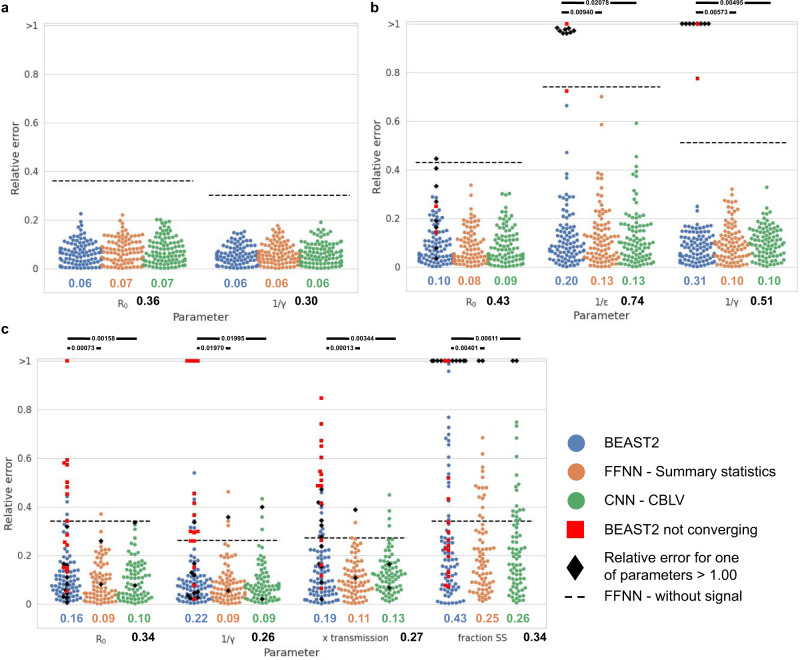
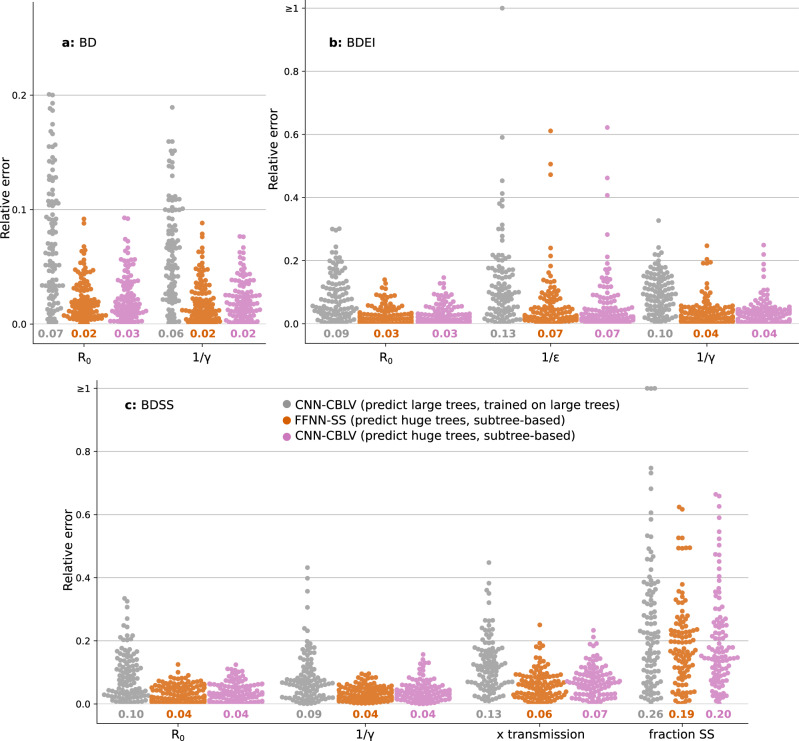
Widely applicable, accurate and fast inference methods in phylodynamics are needed to fully profit from the richness of genetic data in uncovering the dynamics of epidemics. Standard methods, including maximum-likelihood and Bayesian approaches, generally rely on complex mathematical formulae and approximations, and do not scale with dataset size. We develop a likelihood-free, simulation-based approach, which combines deep learning with (1) a large set of summary statistics measured on phylogenies or (2) a complete and compact representation of trees, which avoids potential limitations of summary statistics and applies to any phylodynamics model. Our method enables both model selection and estimation of epidemiological parameters from very large phylogenies. We demonstrate its speed and accuracy on simulated data, where it performs better than the state-of-the-art methods. To illustrate its applicability, we assess the dynamics induced by superspreading individuals in an HIV dataset of men-having-sex-with-men in Zurich. Our tool PhyloDeep is available on github.com/evolbioinfo/phylodeep .
需要在系统发生动力学中广泛应用、准确且快速的推断方法,以充分利用遗传数据的丰富性来揭示流行病的动态。标准方法,包括最大似然法和贝叶斯方法,通常依赖于复杂的数学公式和近似,并且不能随数据集的大小而扩展。我们开发了一种无似然、基于模拟的方法,将深度学习与(1)在系统发生树上测量的大量汇总统计数据或(2)树的完整而紧凑的表示相结合,避免了汇总统计数据的潜在限制,并适用于任何系统发生动力学模型。我们的方法能够从非常大的系统发生树上进行模型选择和流行病学参数的估计。我们在模拟数据上证明了它的速度和准确性,它的性能优于最先进的方法。为了说明其适用性,我们评估了在苏黎世男男性行为者的 HIV 数据集中超级传播者个体引起的动态。我们的工具 PhyloDeep 可在 github.com/evolbioinfo/phylodeep 上获得。