The Second School of Clinical Medicine, Southern Medical University, Guangzhou, China.
Department of Gynecology, Guangdong Second Provincial General Hospital, Guangzhou, China.
Front Cell Infect Microbiol. 2022 Jun 21;12:922554. doi: 10.3389/fcimb.2022.922554. eCollection 2022.
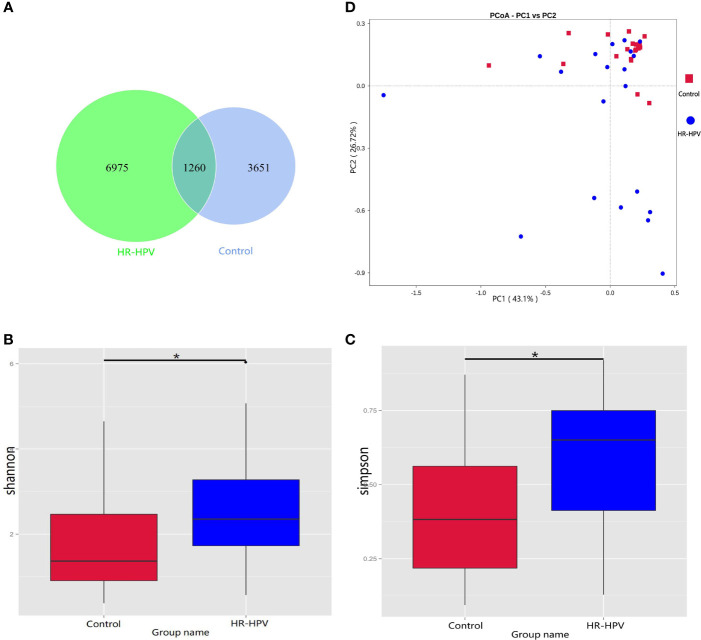
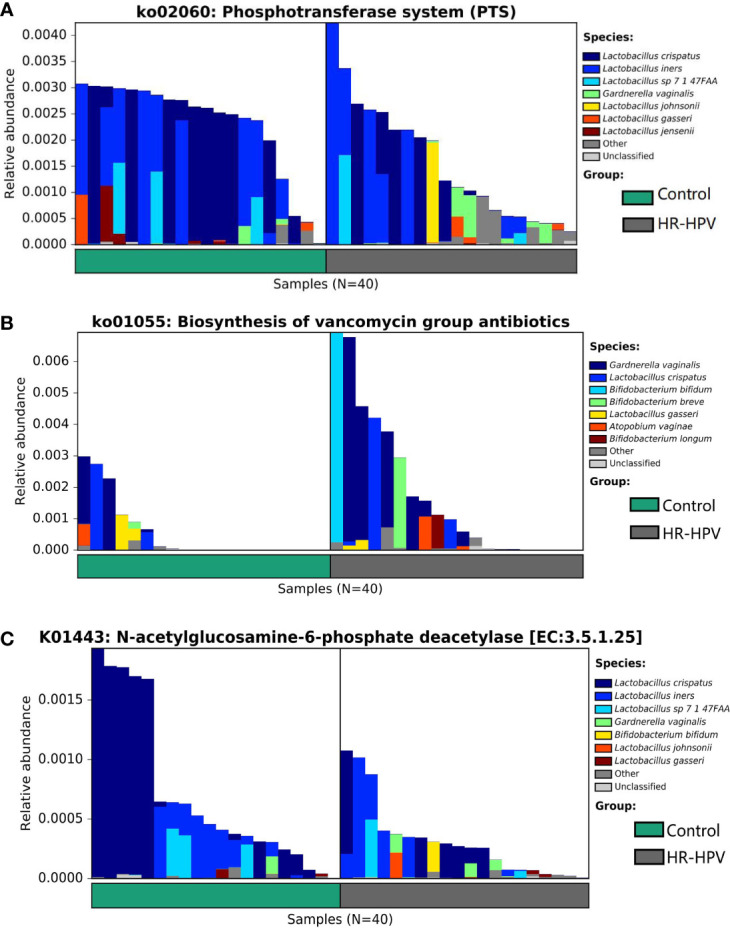
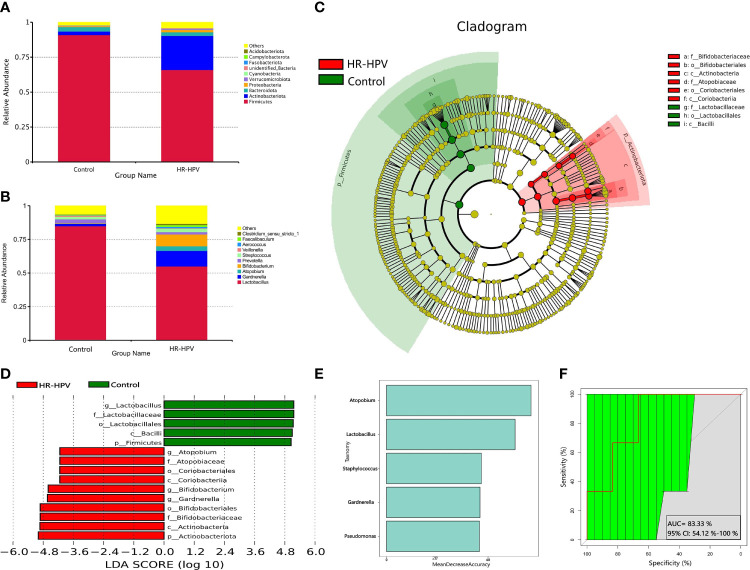
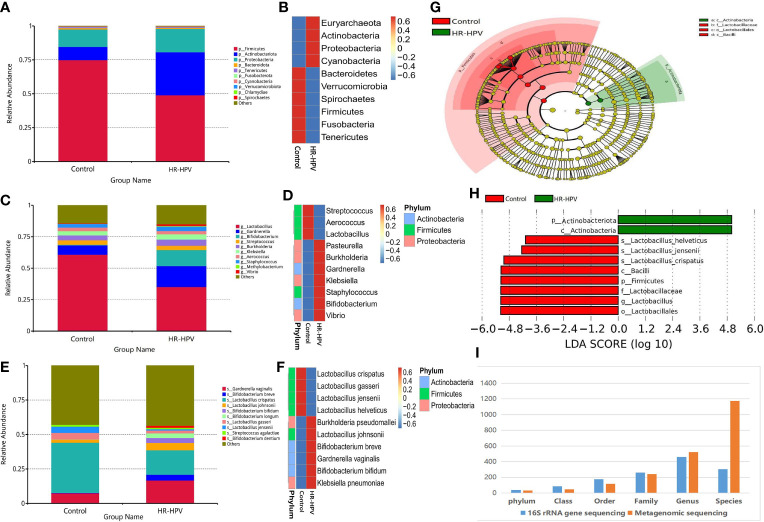
The relationship between the cervico-vaginal microbiome and high-risk human papillomavirus (HR-HPV) is well observed. However, there is a lack of adequate research regarding the cervical microbiota in HR-HPV infection. Most published research results have used 16S rRNA gene sequencing technology; this technology only focuses on marker sequences, resulting in incomplete gene information acquisition. Metagenomic sequencing technology can effectively compensate for the deficiency of 16S rRNA gene sequencing, thus improving the analysis of microbiota function. Cervical swab samples from 20 females with HR-HPV infection and 20 uninfected (Control) women were analyzed through 16S rRNA gene and metagenomic sequencing. Our results indicated that the composition and function of the cervical microbiota of HR-HPV infection differed notably from that of control women. Compared with control women, Firmicutes was decreased during HR-HPV infection, whereas Actinobacteria was increased. At the genus level, Lactobacillus was enriched in control women, while levels of Gardnerella and Bifidobacterium were lower. At the species level, , , and were enriched in control women; these were the top three species with biomarker significance between the two groups. Eight pathways and four KEGG orthologies of the cervical microbiota of statistical differences were identified between the HR-HPV infection and control women. Collectively, our study described the cervical microbiota and its potential function during HR-HPV infection. Biomarkers of cervical microbiota and the changed bacterial metabolic pathways and metabolites can help clarify the pathogenic mechanism of HR-HPV infection, making them promising targets for clinical treatment and intervention for HR-HPV infection and cervical carcinoma.
人乳头瘤病毒(HPV)感染与宫颈微生物群的关系已得到充分观察。然而,关于 HPV 感染宫颈微生物群的研究还不够充分。大多数已发表的研究结果都使用了 16S rRNA 基因测序技术;该技术仅关注标记序列,导致基因信息获取不完整。宏基因组测序技术可以有效地弥补 16S rRNA 基因测序的不足,从而提高对微生物群功能的分析。通过 16S rRNA 基因和宏基因组测序分析了 20 名 HPV 感染的女性和 20 名未感染(对照)女性的宫颈拭子样本。我们的结果表明,HPV 感染女性的宫颈微生物群组成和功能与对照组女性明显不同。与对照组女性相比,HPV 感染期间厚壁菌门减少,而放线菌门增加。在属水平上,对照组女性中乳杆菌丰富,而加德纳菌和双歧杆菌水平较低。在种水平上, 、 、 在对照组女性中富集;这是两组之间具有生物标志物意义的前三种物种。HPV 感染和对照组女性之间的宫颈微生物群有 8 条途径和 4 个 KEGG 直系同源物存在统计学差异。总之,本研究描述了 HPV 感染期间宫颈微生物群及其潜在功能。宫颈微生物群的生物标志物和改变的细菌代谢途径和代谢物有助于阐明 HPV 感染的致病机制,使它们成为 HPV 感染和宫颈癌临床治疗和干预的有前途的靶点。