Mukaidaisi Muhetaer, Vu Andrew, Grantham Karl, Tchagang Alain, Li Yifeng
Biomedical Data Science Laboratory, Department of Computer Science, Brock University, St. Catharines, ON, Canada.
Scientific Data Mining Team, Digital Technologies Research Centre, National Research Council Canada, Ottawa, ON, Canada.
Front Pharmacol. 2022 Jul 4;13:920747. doi: 10.3389/fphar.2022.920747. eCollection 2022.
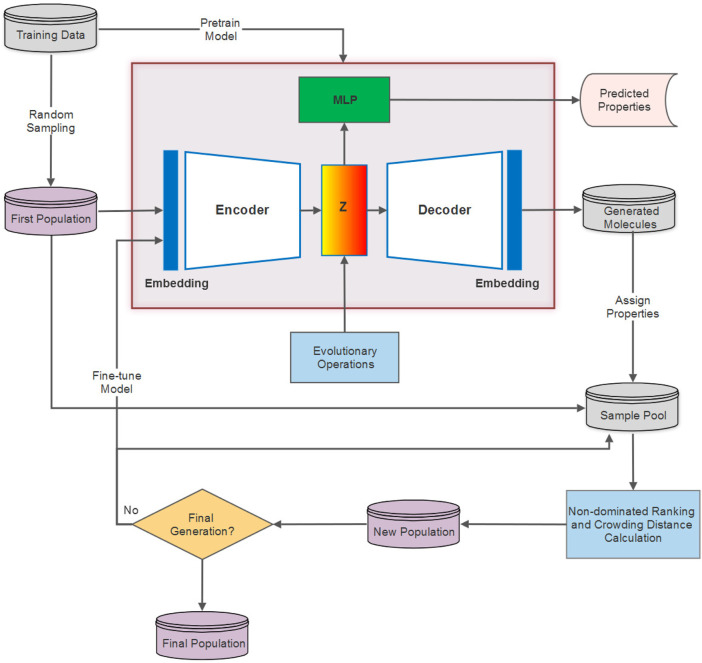
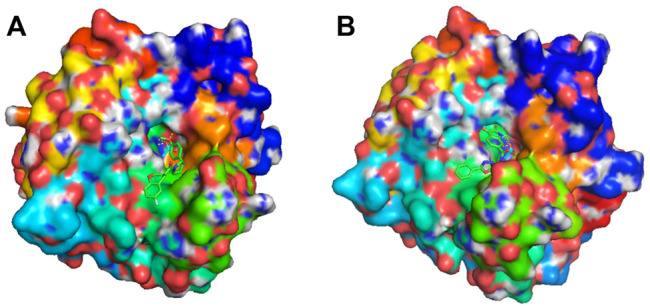
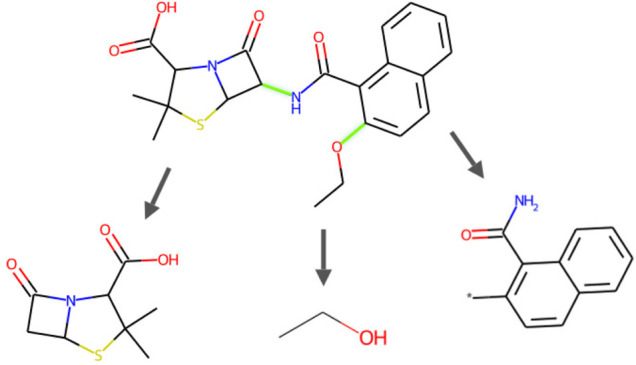
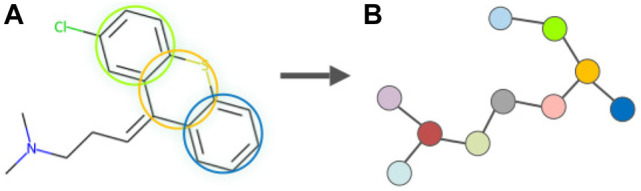
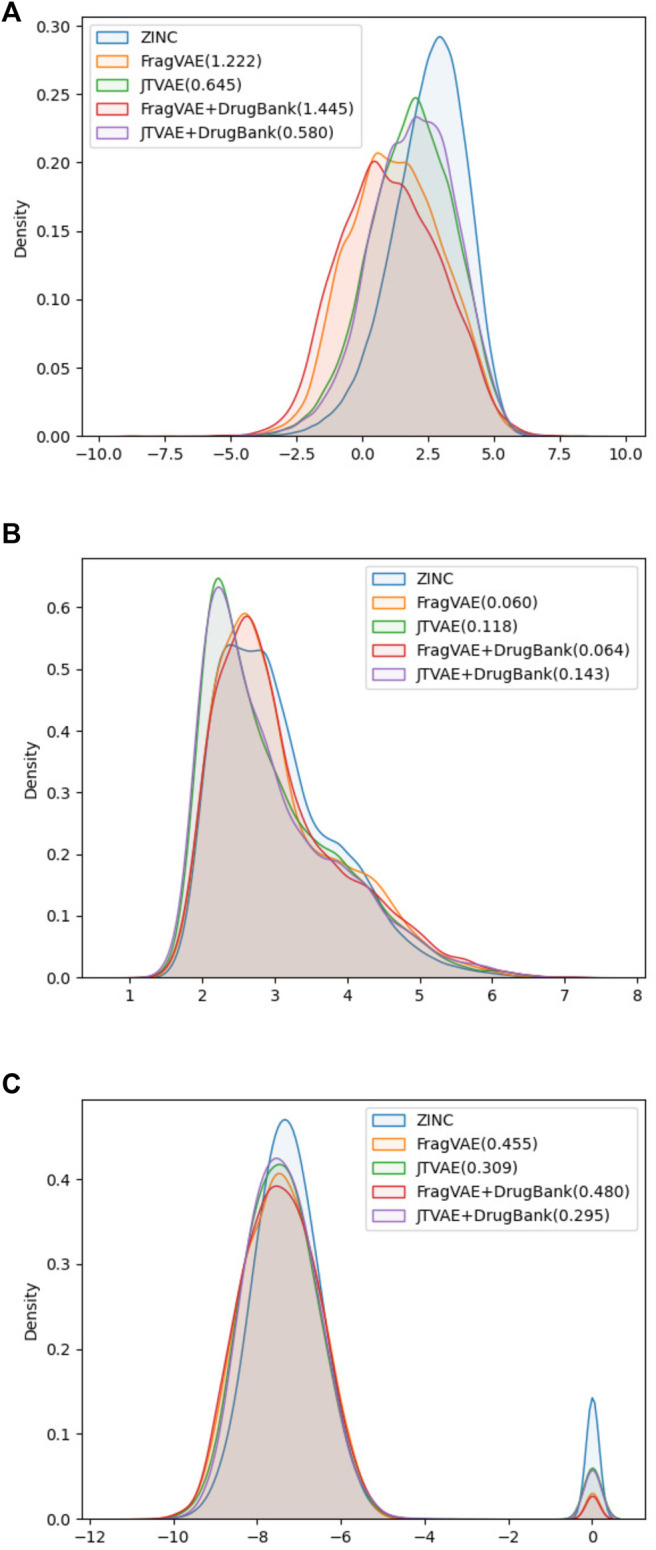
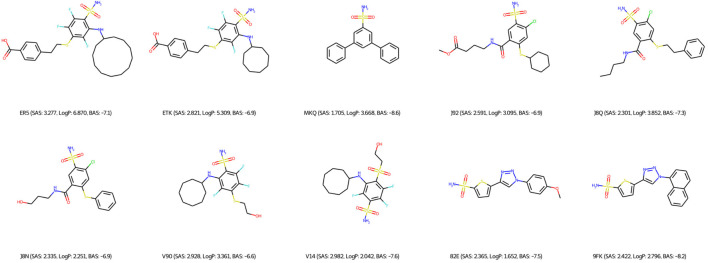
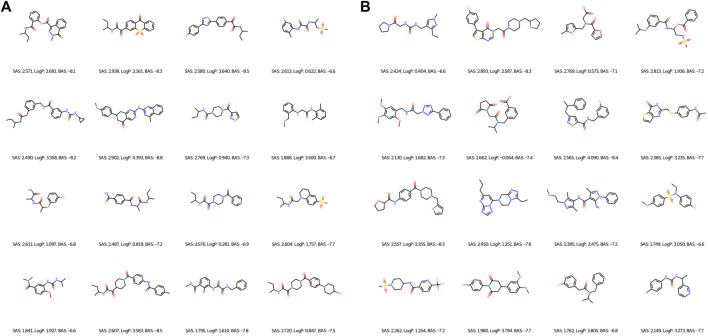
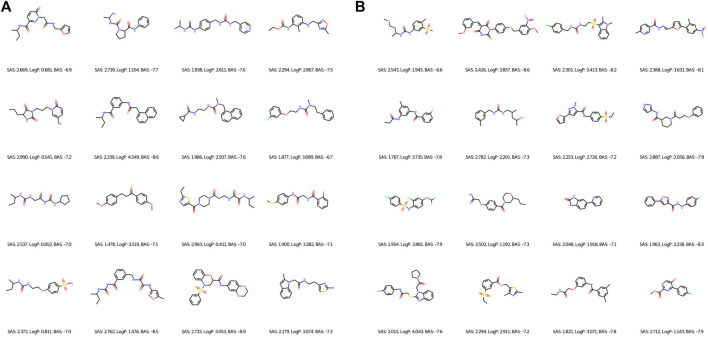
Drug discovery is a challenging process with a huge molecular space to be explored and numerous pharmacological properties to be appropriately considered. Among various drug design protocols, fragment-based drug design is an effective way of constraining the search space and better utilizing biologically active compounds. Motivated by fragment-based drug search for a given protein target and the emergence of artificial intelligence (AI) approaches in this field, this work advances the field of drug design by (1) integrating a graph fragmentation-based deep generative model with a deep evolutionary learning process for large-scale multi-objective molecular optimization, and (2) applying protein-ligand binding affinity scores together with other desired physicochemical properties as objectives. Our experiments show that the proposed method can generate novel molecules with improved property values and binding affinities.
药物发现是一个具有挑战性的过程,需要探索巨大的分子空间,并适当考虑众多药理特性。在各种药物设计方案中,基于片段的药物设计是一种有效限制搜索空间并更好利用生物活性化合物的方法。受针对给定蛋白质靶点的基于片段的药物搜索以及该领域人工智能(AI)方法出现的启发,本研究通过以下方式推动了药物设计领域的发展:(1)将基于图片段的深度生成模型与深度进化学习过程相结合,用于大规模多目标分子优化;(2)将蛋白质-配体结合亲和力得分与其他所需的物理化学性质一起作为目标。我们的实验表明,所提出的方法可以生成具有改进性质值和结合亲和力的新型分子。