White Jon, Sharma Rajiv, Balding David, Cockram James, Mackay Ian J
Genetics and Breeding Dep. NIAB 93 Lawrence Weaver Road Cambridge, CB3 0LE UK.
Institute of Genetics Univ. College London London, WC1E 6BT UK.
Crop Sci. 2022 May-Jun;62(3):965-981. doi: 10.1002/csc2.20692. Epub 2022 Mar 4.
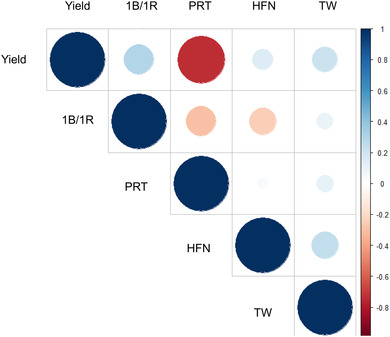
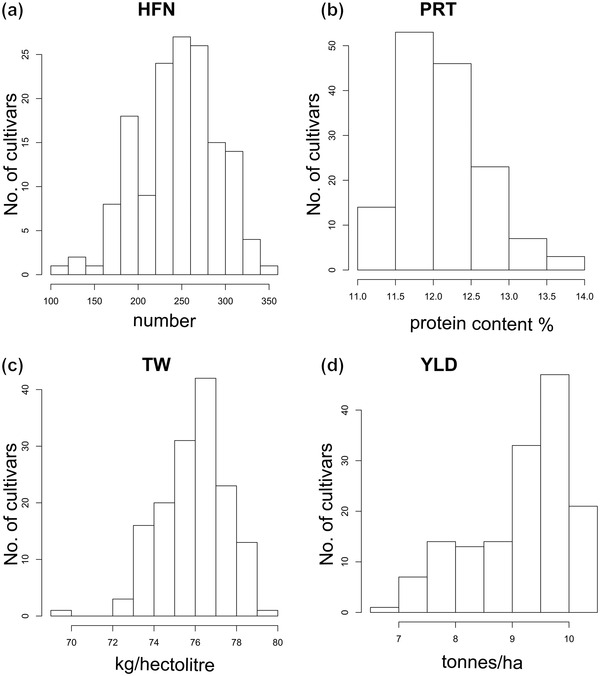
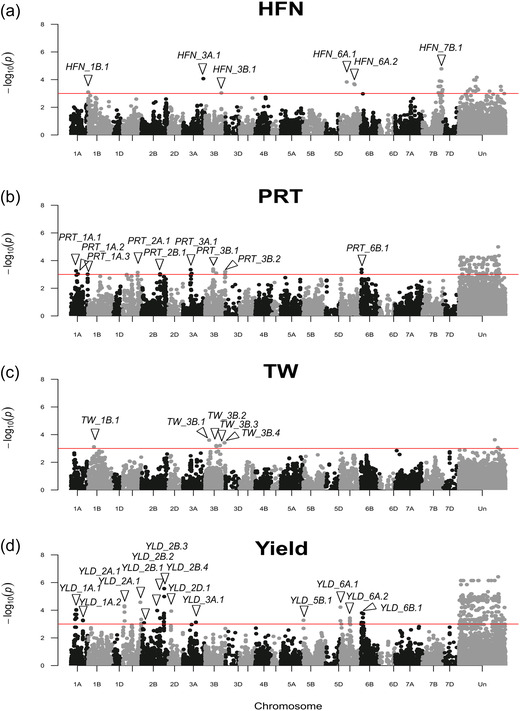
Association mapping using crop cultivars allows identification of genetic loci of direct relevance to breeding. Here, 150 U.K. wheat ( L.) cultivars genotyped with 23,288 single nucleotide polymorphisms (SNPs) were used for genome-wide association studies (GWAS) using historical phenotypic data for grain protein content, Hagberg falling number (HFN), test weight, and grain yield. Power calculations indicated experimental design would enable detection of quantitative trait loci (QTL) explaining ≥20% of the variation (PVE) at a relatively high power of >80%, falling to 40% for detection of a SNP with an ≥ .5 with the same QTL. Genome-wide association studies identified marker-trait associations for all four traits. For HFN ( = .89), six QTL were identified, including a major locus on chromosome 7B explaining 49% PVE and reducing HFN by 44 s. For protein content ( = 0.86), 10 QTL were found on chromosomes 1A, 2A, 2B, 3A, 3B, and 6B, together explaining 48.9% PVE. For test weight, five QTL were identified (one on 1B and four on 3B; 26.3% PVE). Finally, 14 loci were identified for grain yield ( = 0.95) on eight chromosomes (1A, 2A, 2B, 2D, 3A, 5B, 6A, 6B; 68.1% PVE), of which five were located within 16 Mbp of genetic regions previously identified as under breeder selection in European wheat. Our study demonstrates the utility of exploiting historical crop datasets, identifying genomic targets for independent validation, and ultimately for wheat genetic improvement.
利用作物品种进行关联作图有助于识别与育种直接相关的基因座。在此,对150个英国小麦(L.)品种进行了基因分型,共检测到23288个单核苷酸多态性(SNP),并利用历史表型数据进行全基因组关联研究(GWAS),这些表型数据包括籽粒蛋白质含量、哈格伯格沉降值(HFN)、容重和籽粒产量。功效计算表明,实验设计能够以相对较高的功效(>80%)检测到解释≥20%变异(PVE)的数量性状基因座(QTL),对于检测具有相同QTL且PVE≥0.5的SNP,功效降至40%。全基因组关联研究确定了所有四个性状的标记-性状关联。对于HFN(r = 0.89),鉴定出6个QTL,包括7B染色体上的一个主效基因座,解释了49%的PVE,并使HFN降低44秒。对于蛋白质含量(r = 0.86),在1A、2A、2B、3A、3B和6B染色体上发现了10个QTL,共同解释了48.9%的PVE。对于容重,鉴定出5个QTL(1个在1B染色体上,4个在3B染色体上;26.3%的PVE)。最后,在8条染色体(1A、2A、2B、2D、3A、5B、6A、6B;68.1%的PVE)上鉴定出14个籽粒产量基因座,其中5个位于先前在欧洲小麦育种选择中确定的遗传区域的16 Mbp范围内。我们的研究证明了利用历史作物数据集、识别用于独立验证的基因组靶点以及最终用于小麦遗传改良的实用性。