Epasto Ludovica M, Che Kateryna, Kozak Fanny, Selimovic Albina, Kadeřávek Pavel, Kurzbach Dennis
University of Vienna, Faculty of Chemistry, Institute of Biological Chemistry, Währinger Str. 38, 1090 Vienna, Austria.
Masaryk University, CEITEC, Kamenice 5, 625 00 Brno, Czech Republic.
Sci Adv. 2022 Aug 5;8(31):eabq5179. doi: 10.1126/sciadv.abq5179.
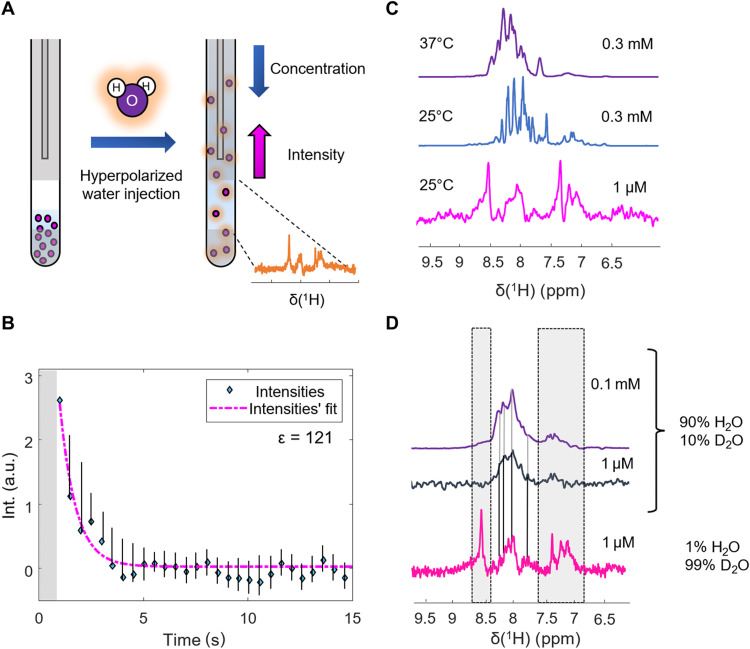
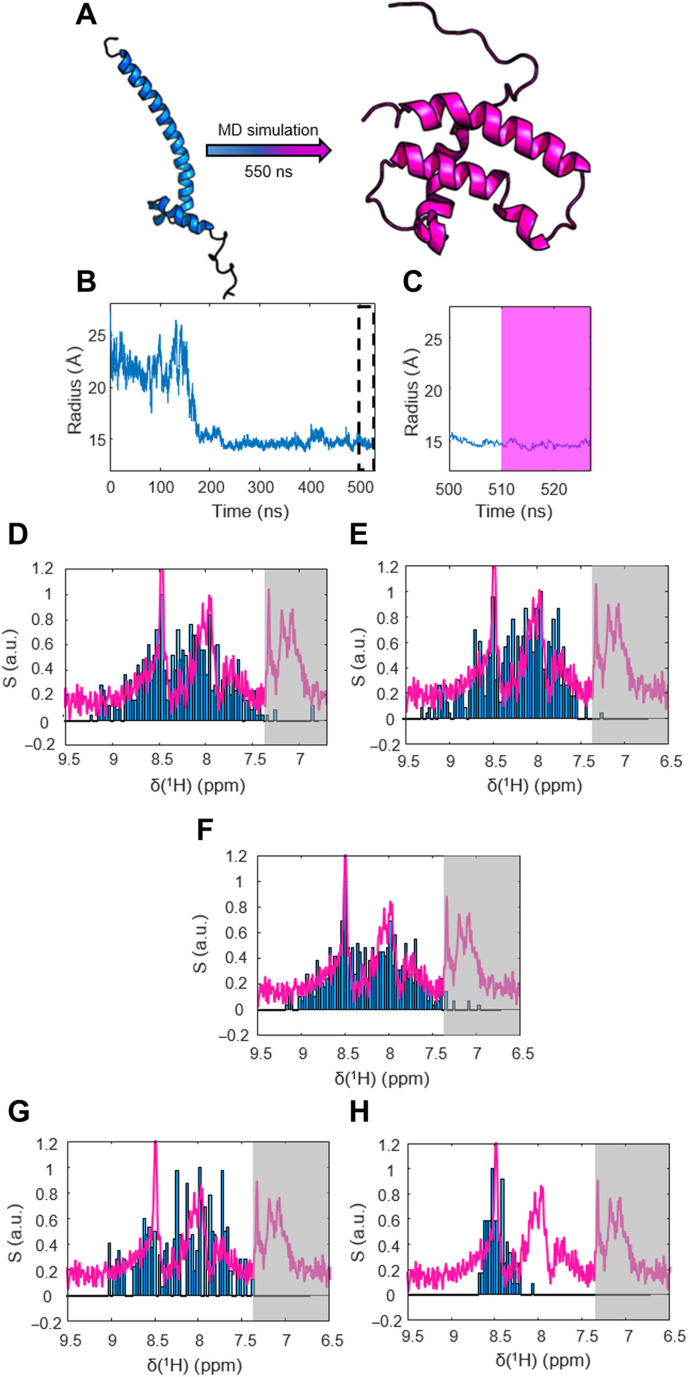
Nuclear magnetic resonance (NMR) spectroscopy is a key method for determining the structural dynamics of proteins in their native solution state. However, the low sensitivity of NMR typically necessitates nonphysiologically high sample concentrations, which often limit the relevance of the recorded data. We show how to use hyperpolarized water by dissolution dynamic nuclear polarization (DDNP) to acquire protein spectra at concentrations of 1 μM within seconds and with a high signal-to-noise ratio. The importance of approaching physiological concentrations is demonstrated for the vital MYC-associated factor X, which we show to switch conformations when diluted. While in vitro conditions lead to a population of the well-documented dimer, concentrations lowered by more than two orders of magnitude entail dimer dissociation and formation of a globularly folded monomer. We identified this structure by integrating DDNP with computational techniques to overcome the often-encountered constraint of DDNP of limited structural information provided by the typically detected one-dimensional spectra.
核磁共振(NMR)光谱法是确定蛋白质在其天然溶液状态下结构动力学的关键方法。然而,NMR的低灵敏度通常需要非生理状态下的高样品浓度,这常常限制了所记录数据的相关性。我们展示了如何通过溶解动态核极化(DDNP)使用超极化水,在几秒钟内以1 μM的浓度获取蛋白质光谱,且具有高信噪比。对于至关重要的MYC相关因子X,接近生理浓度的重要性得到了证明,我们发现当稀释时它会发生构象转变。虽然体外条件会导致形成大量文献记载的二聚体,但浓度降低两个以上数量级会导致二聚体解离并形成球状折叠的单体。我们通过将DDNP与计算技术相结合来识别这种结构,以克服通常检测到的一维光谱所提供的有限结构信息这一DDNP经常遇到的限制。