Markovich Zachary R, Hartman Jessica H, Ryde Ian T, Hershberger Kathleen A, Joyce Abigail S, Ferguson Patrick L, Meyer Joel N
Nicholas School of the Environment, Duke University, Durham, NC 27708-0328, USA.
Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC 29425, USA.
Curr Res Toxicol. 2022 Aug 2;3:100084. doi: 10.1016/j.crtox.2022.100084. eCollection 2022.
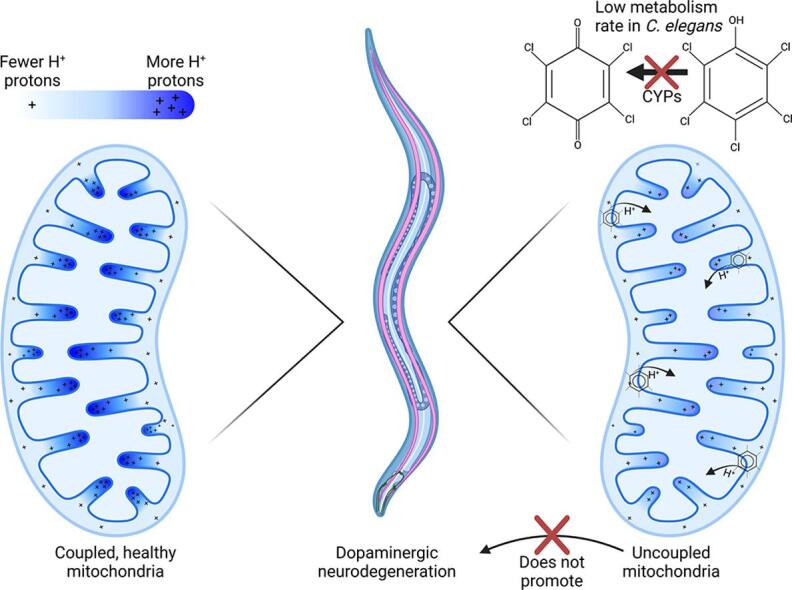
Mitochondrial dysfunction is implicated in several diseases, including neurological disorders such as Parkinson's disease. However, there is uncertainty about which of the many mechanisms by which mitochondrial function can be disrupted may lead to neurodegeneration. Pentachlorophenol (PCP) is an organic pollutant reported to cause mitochondrial dysfunction including oxidative stress and mitochondrial uncoupling. We investigated the effects of PCP exposure in , including effects on mitochondria and dopaminergic neurons. We hypothesized that mild mitochondrial uncoupling by PCP would impair bioenergetics while decreasing oxidative stress, and therefore would not cause dopaminergic neurodegeneration.
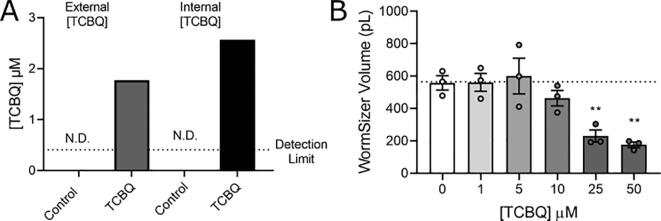
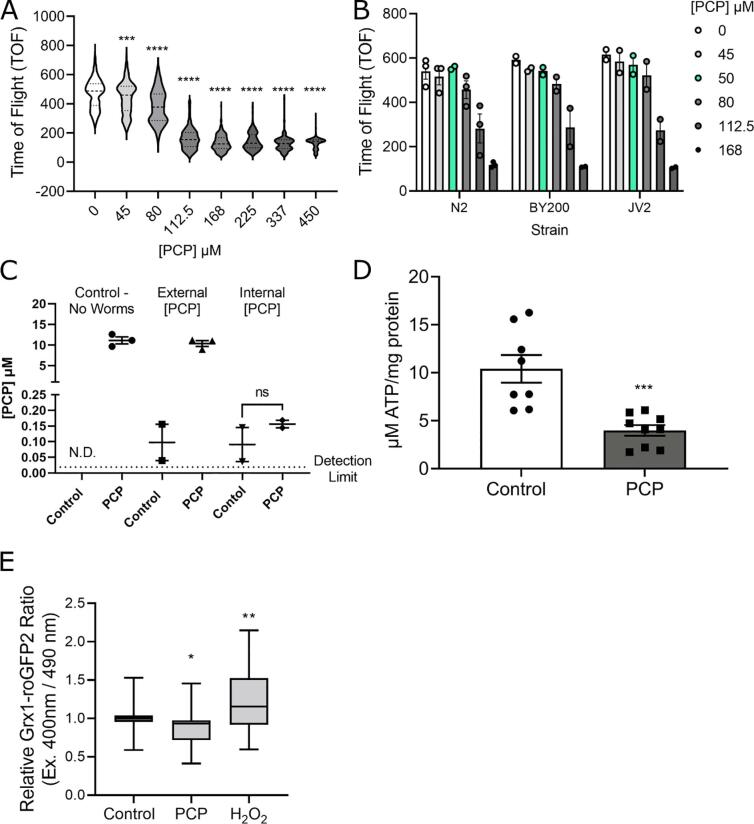
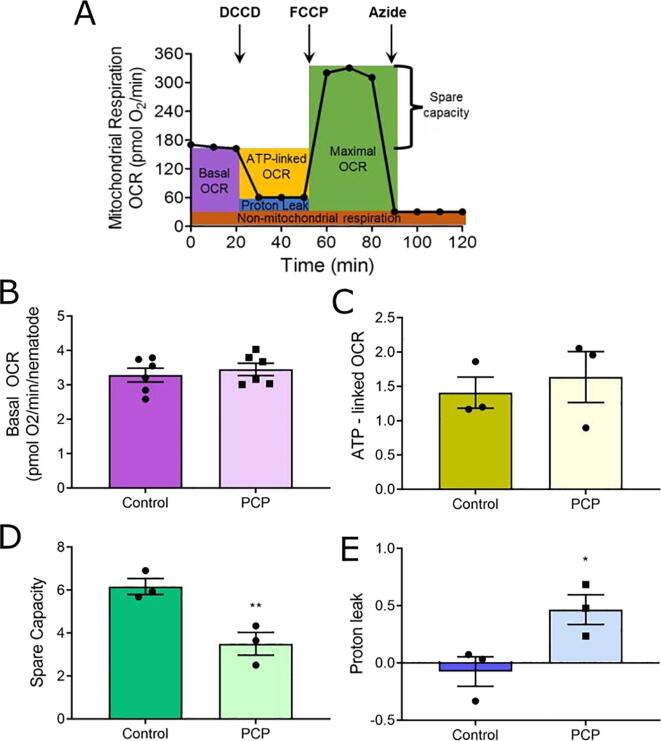
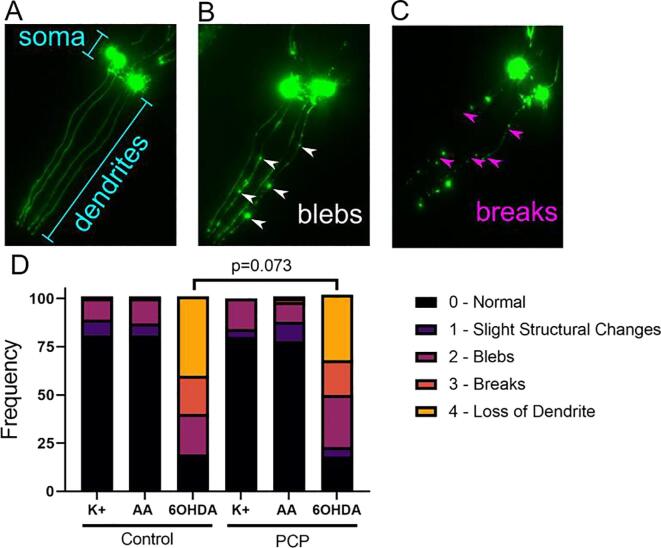
A 48-hour developmental exposure to PCP causing mild growth delay (∼10 % decrease in growth during 48 h, covering all larval stages) reduced whole-organism ATP content > 50 %, and spare respiratory capacity ∼ 30 %. Proton leak was also markedly increased. These findings suggest a main toxic mechanism of mitochondrial uncoupling rather than oxidative stress, which was further supported by a concomitant shift toward a more reduced cellular redox state measured at the whole organism level. However, exposure to PCP did not cause dopaminergic neurodegeneration, nor did it sensitize animals to a neurotoxic challenge with 6-hydroxydopamine. Whole-organism uptake and PCP metabolism measurements revealed low overall uptake of PCP in our experimental conditions (50 μM PCP in the liquid exposure medium resulted in organismal concentrations of < 0.25 μM), and no measurable production of the oxidative metabolites tetra-1,4-benzoquinone and tetrachloro-p-hydroquinone.
This study provides new insights into the mechanistic interplay between mitochondrial uncoupling, oxidative stress, and neurodegeneration in These findings support the premise of mild uncoupling-mediated neuroprotection, but are inconsistent with proposed broad "mitochondrial dysfunction"-mediated neurodegeneration models, and highlight the utility of the model for studying mitochondrial and neurotoxicity.
Developmental exposure to pentachlorophenol causes gross toxicological effects (growth delay and arrest) at high levels. At a lower level of exposure, still causing mild growth delay, we observed mitochondrial dysfunction including uncoupling and decreased ATP levels. However, this was associated with a more-reduced cellular redox tone and did not exacerbate dopaminergic neurotoxicity of 6-hydroxydopamine, instead trending toward protection. These findings may be informative of efforts to define nuanced mitochondrial dysfunction-related adverse outcome pathways that will differ depending on the form of initial mitochondrial toxicity.
线粒体功能障碍与多种疾病有关,包括帕金森病等神经疾病。然而,线粒体功能可能被破坏的众多机制中,究竟哪一种会导致神经退行性变尚不确定。五氯苯酚(PCP)是一种有机污染物,据报道可导致线粒体功能障碍,包括氧化应激和线粒体解偶联。我们研究了PCP暴露对[具体对象未明确]的影响,包括对线粒体和多巴胺能神经元的影响。我们假设PCP引起的轻度线粒体解偶联会损害生物能量学,同时降低氧化应激,因此不会导致多巴胺能神经退行性变。
48小时的发育期PCP暴露导致轻度生长延迟(48小时内生长减少约10%,涵盖所有幼虫阶段),使全生物体ATP含量降低>50%,备用呼吸能力降低约30%。质子泄漏也明显增加。这些发现提示线粒体解偶联而非氧化应激是主要毒性机制,这在全生物体水平上同时向更还原的细胞氧化还原状态转变中得到进一步支持。然而,PCP暴露并未导致多巴胺能神经退行性变,也未使动物对6-羟基多巴胺的神经毒性刺激敏感。全生物体对PCP的摄取和PCP代谢测量显示,在我们的实验条件下PCP的总体摄取量较低(液体暴露介质中50μM的PCP导致生物体浓度<0.25μM),且未检测到氧化代谢产物四氯-1,4-苯醌和四氯对苯二酚的产生。
本研究为[具体对象未明确]中线粒体解偶联、氧化应激和神经退行性变之间的机制相互作用提供了新见解。这些发现支持轻度解偶联介导的神经保护前提,但与所提出的广泛的“线粒体功能障碍”介导的神经退行性变模型不一致,并突出了[具体对象未明确]模型在研究线粒体和神经毒性方面的实用性。
发育期暴露于五氯苯酚在高剂量时会导致严重的毒理学效应(生长延迟和停滞)。在较低暴露水平下,仍会导致轻度生长延迟,我们观察到线粒体功能障碍,包括解偶联和ATP水平降低。然而,这与更还原的细胞氧化还原状态相关,且并未加剧6-羟基多巴胺对多巴胺能的神经毒性,反而有保护趋势。这些发现可能有助于确定与线粒体功能障碍相关的细微不良结局途径,这些途径会因初始线粒体毒性的形式不同而有所差异。