Xu Jie, Wang Jinghuan, Long Fen, Zhong Wen, Su Haibi, Su Zhenghua, Liu Xinhua
School of Pharmacy, Pharmacophenomics Laboratory, Human Phenome Institute, Fudan University, 825, Zhangheng Road, Pudong New District, Shanghai, 201203, People's Republic of China.
Cell Biosci. 2022 Aug 19;12(1):134. doi: 10.1186/s13578-022-00877-5.
Cardiac fibrosis is characterized by excessive extracellular matrix deposition that contributes to compromised cardiac function and potentially heart failure. Disruptor of telomeric silencing 1-like (Dot1L) is the catalytic enzyme required for histone H3K79 methylation which has been demonstrated to play a role in transcriptional activation. However, the functions of Dot1L in the process of cardiac fibrosis still remain unknown.
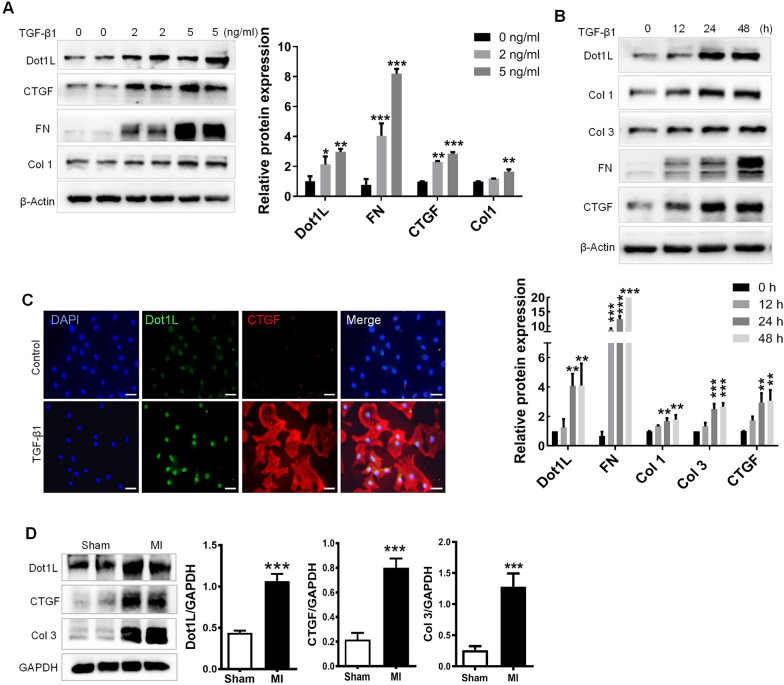
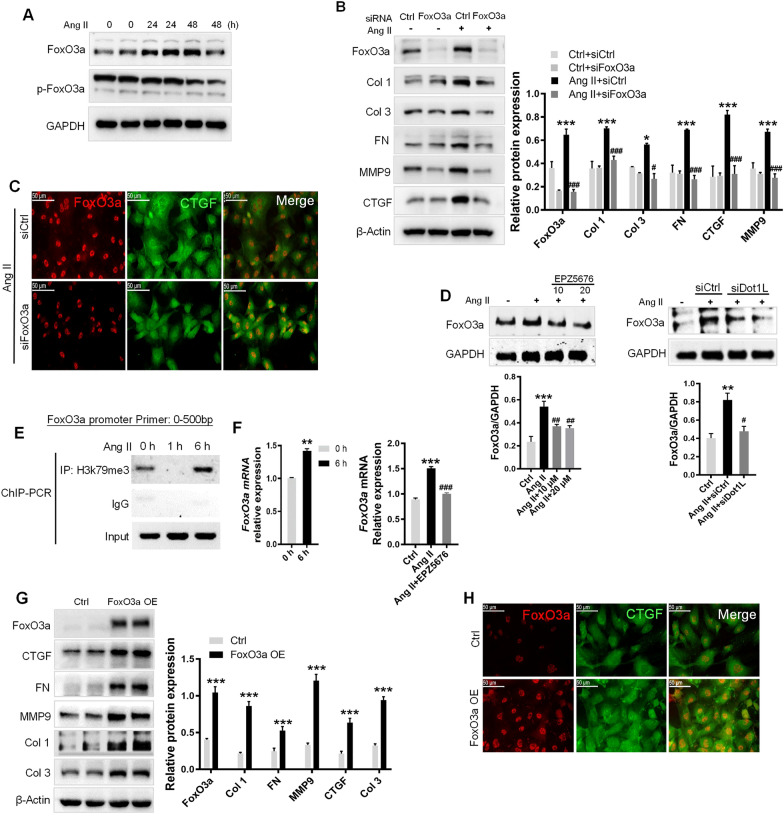
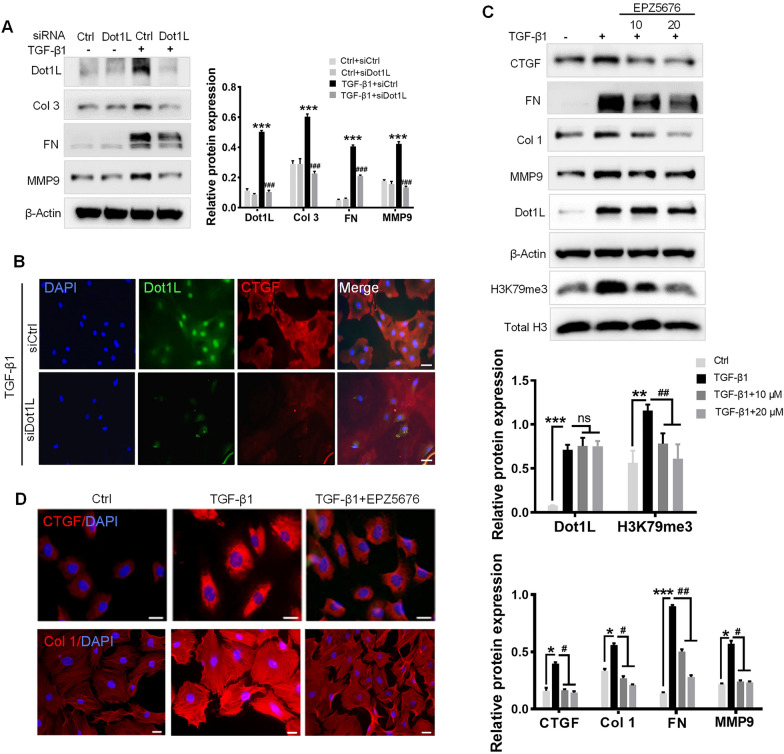
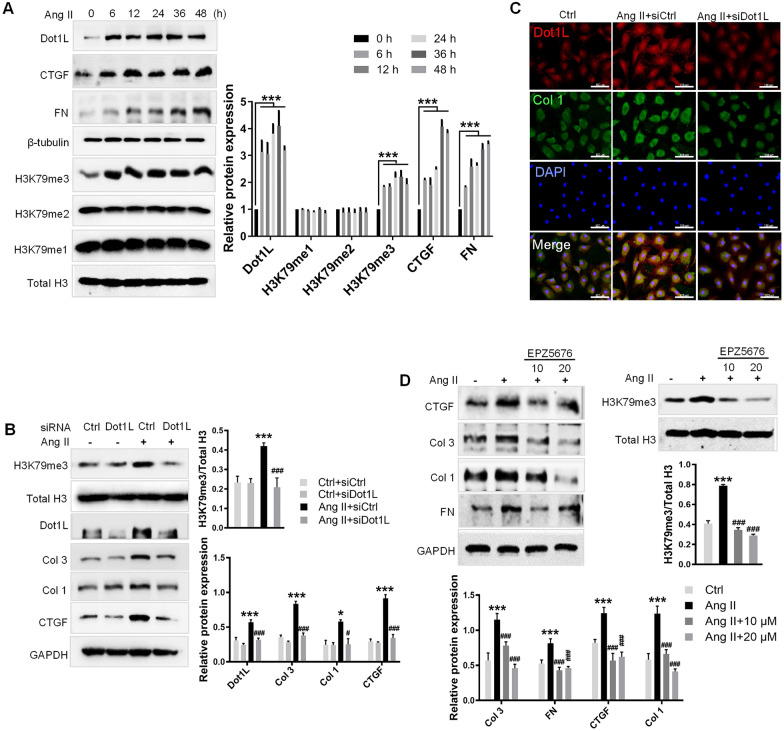
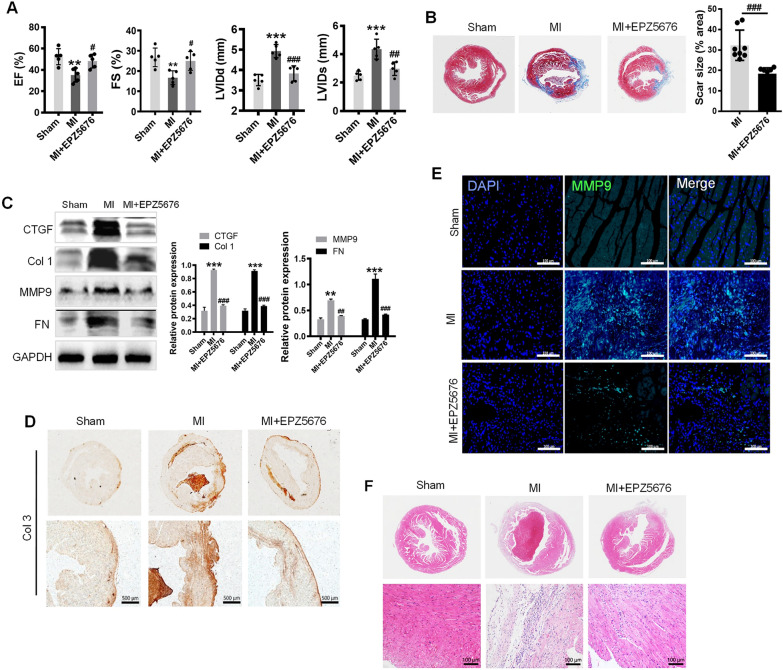
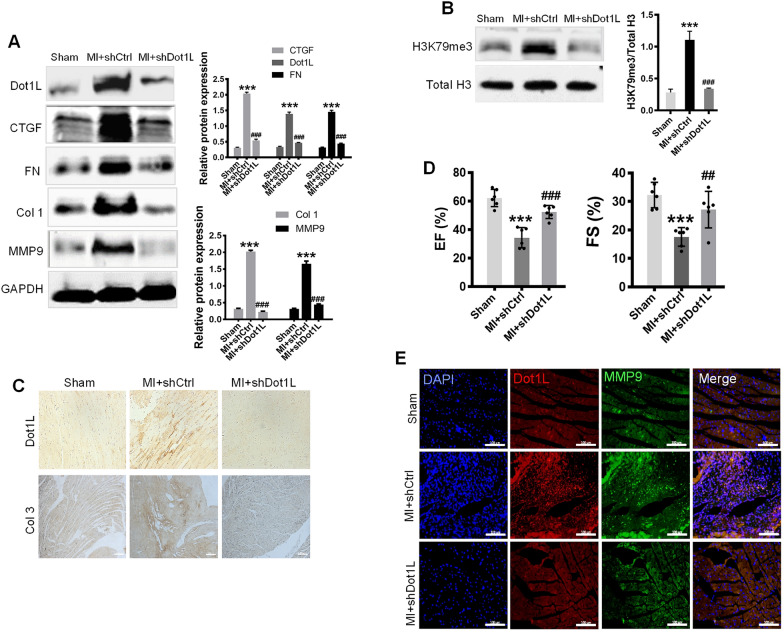
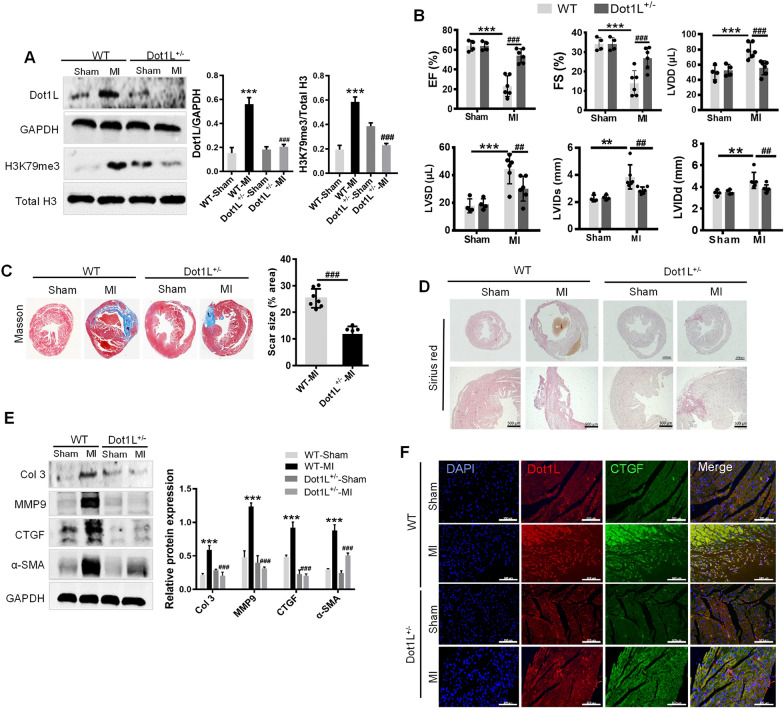
In the present study, we found that endogenous Dot1L is upregulated in cardiac fibroblasts (CFs) treated with angiotensin II (Ang II) or transforming growth factor (TGF)-β1, along with elevated extracellular matrix (ECM) such as fibronectin, collagen I and III. Silencing or inhibiting Dot1L mitigated Ang II-induced myofibroblast generation and fibrogenesis. We identified the transcription factor-forkhead box O (FoxO) 3a as a novel substrate of Dot1L, the transcriptional activating mark H3K79me3 level on the promoter of FoxO3a was increase in activated-CFs, and inhibition of Dot1L markedly decreased FoxO3a transcription accompanied by a significant decrease in the expression of fibrogenic gene. Knockdown of FoxO3a could alleviate ECM deposition induced by Ang II, on the contrary, overexpression FoxO3a resulting in CFs activation. Consistently, in vivo Dot1L ablation rescued myocardial ischemia-induced cardiac fibrosis and improved cardiac function.
Our findings conclude that upregulation of Dot1L results in activation of the cardiac fibroblasts to promote profibrotic gene, eventually causes cardiac fibrosis. Pharmacological targeting for Dot1L might represent a promising therapeutic approach for the treatment of human cardiac fibrosis and other fibrotic diseases.
心脏纤维化的特征是细胞外基质过度沉积,这会导致心脏功能受损并可能引发心力衰竭。端粒沉默破坏因子1样蛋白(Dot1L)是组蛋白H3K79甲基化所需的催化酶,已证明其在转录激活中发挥作用。然而,Dot1L在心脏纤维化过程中的功能仍不清楚。
在本研究中,我们发现用血管紧张素II(Ang II)或转化生长因子(TGF)-β1处理的心脏成纤维细胞(CFs)中内源性Dot1L上调,同时细胞外基质(ECM)如纤连蛋白、I型和III型胶原也升高。沉默或抑制Dot1L可减轻Ang II诱导的肌成纤维细胞生成和纤维化。我们确定转录因子叉头框O(FoxO)3a是Dot1L的一种新底物,在活化的CFs中FoxO3a启动子上的转录激活标记H3K79me3水平升高,抑制Dot1L可显著降低FoxO3a转录,并伴随纤维化基因表达的显著降低。敲低FoxO3a可减轻Ang II诱导的ECM沉积,相反,过表达FoxO3a会导致CFs活化。同样,在体内,Dot1L基因敲除可挽救心肌缺血诱导的心脏纤维化并改善心脏功能。
我们的研究结果表明,Dot1L上调导致心脏成纤维细胞活化,促进促纤维化基因表达,最终导致心脏纤维化。针对Dot1L的药物靶向治疗可能是治疗人类心脏纤维化和其他纤维化疾病的一种有前景的治疗方法。