Wang Zhen, Zhang Zhiyuan, Zhang Chen, Jin Xin, Wu Jianjun, Su Bin, Shen Yuelan, Ruan Yuhua, Xing Hui, Lou Jie
Department of Mathematics, Shanghai University, Shanghai 200444, China.
Department of Statistics, Columbia University, New York, NY 10027, USA.
Trop Med Infect Dis. 2022 Aug 17;7(8):190. doi: 10.3390/tropicalmed7080190.
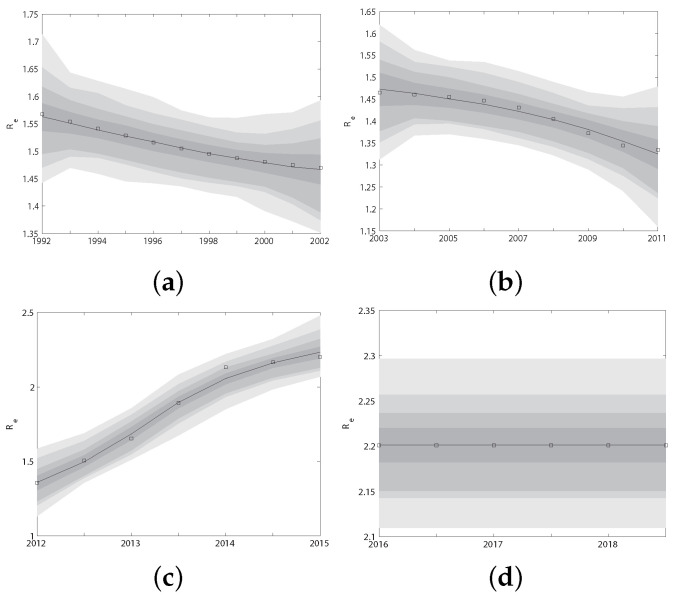
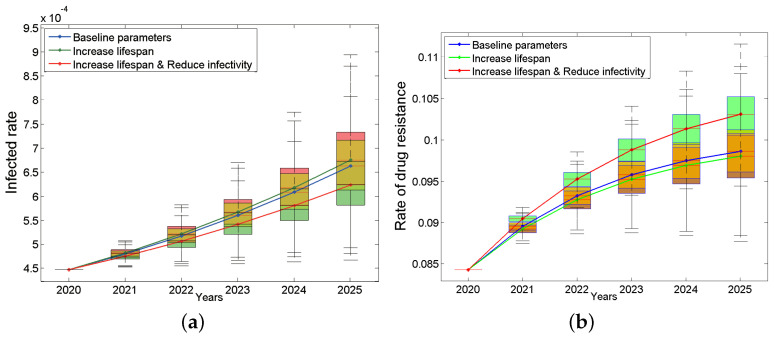
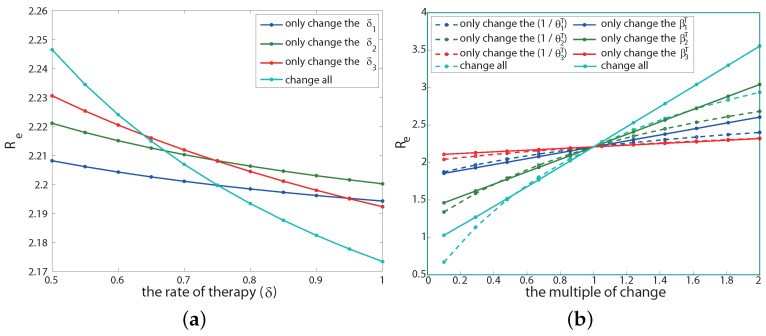
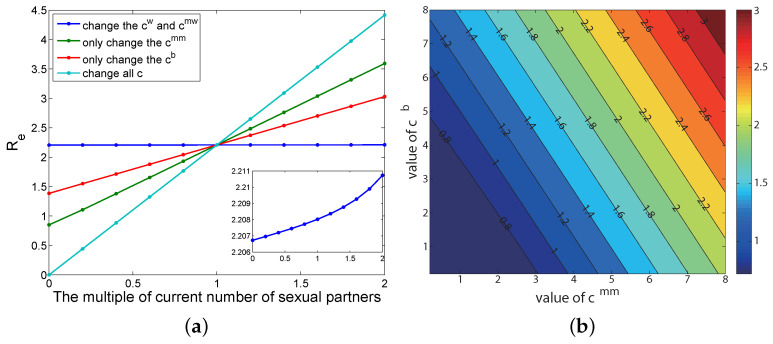
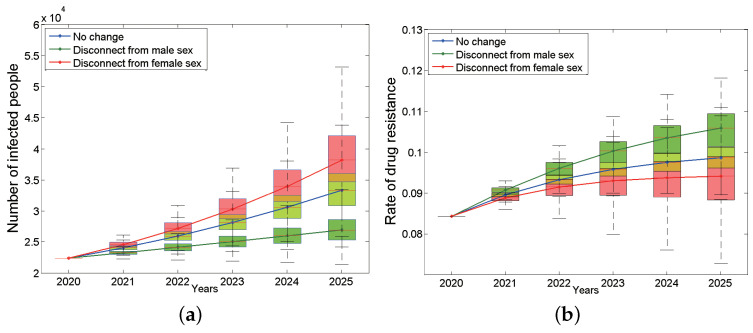
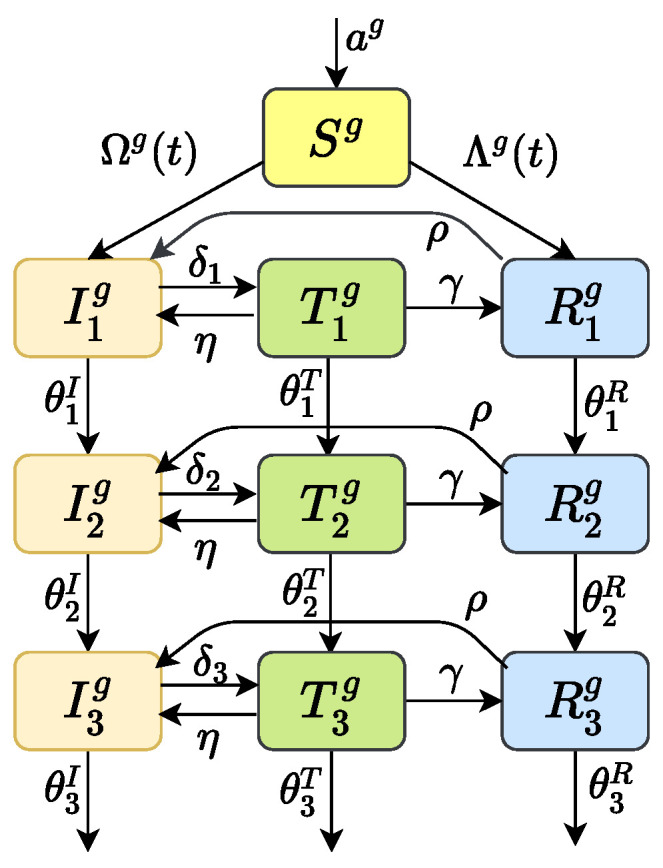
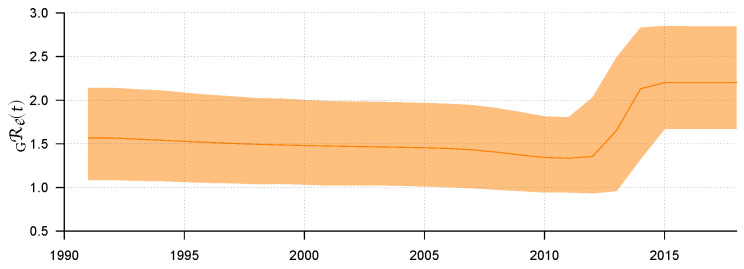
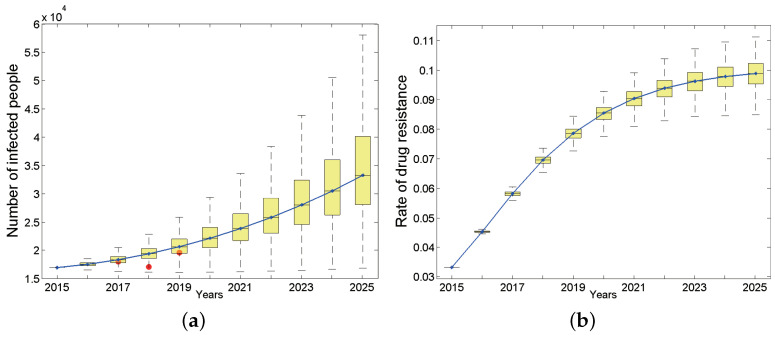
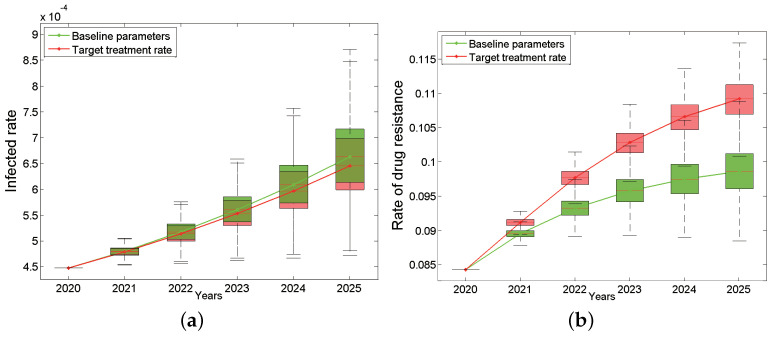
Traditional methods of quantifying epidemic spread are based on surveillance data. The most widely used surveillance data are normally incidence data from case reports and hospital records, which are normally susceptible to human error, and sometimes, they even can be seriously error-prone and incomplete when collected during a destructive epidemic. In this manuscript, we introduce a new method to study the spread of infectious disease. We gave an example of how to use this method to predict the virus spreading using the HIV gene sequences data of China. First, we applied Bayesian inference to gene sequences of two main subtypes of the HIV virus to infer the effective reproduction number (GRe(t)) to trace the history of HIV transmission. Second, a dynamic model was established to forecast the spread of HIV medication resistance in the future and also obtain its effective reproduction number (MRe(t)). Through fitting the two effective reproduction numbers obtained from the two separate ways above, some crucial parameters for the dynamic model were obtained. Simply raising the treatment rate has no impact on lowering the infection rate, according to the dynamics model research, but would instead increase the rate of medication resistance. The negative relationship between the prevalence of HIV and the survivorship of infected individuals following treatment may be to blame for this. Reducing the MSM population's number of sexual partners is a more efficient strategy to reduce transmission per the sensitivity analysis.
传统的量化疫情传播的方法基于监测数据。最广泛使用的监测数据通常是病例报告和医院记录中的发病率数据,这些数据通常容易出现人为错误,而且在破坏性疫情期间收集时,有时甚至可能极易出错且不完整。在本论文中,我们介绍一种研究传染病传播的新方法。我们给出了一个如何使用该方法利用中国的HIV基因序列数据预测病毒传播的例子。首先,我们对HIV病毒的两个主要亚型的基因序列应用贝叶斯推断来推断有效再生数(GRe(t))以追溯HIV传播的历史。其次,建立一个动态模型来预测未来HIV耐药性的传播,并获得其有效再生数(MRe(t))。通过拟合从上述两种不同方法获得的两个有效再生数,得到了动态模型的一些关键参数。根据动力学模型研究,单纯提高治疗率对降低感染率没有影响,反而会增加耐药率。这可能归咎于HIV患病率与治疗后感染者存活率之间的负相关关系。根据敏感性分析,减少男男性行为人群的性伴侣数量是降低传播的更有效策略。