Department of Cell & Molecular Biology, Uppsala University, Biomedical Center, SE-751 24 Uppsala, Sweden.
J Chem Theory Comput. 2022 Oct 11;18(10):6345-6353. doi: 10.1021/acs.jctc.2c00646. Epub 2022 Sep 12.
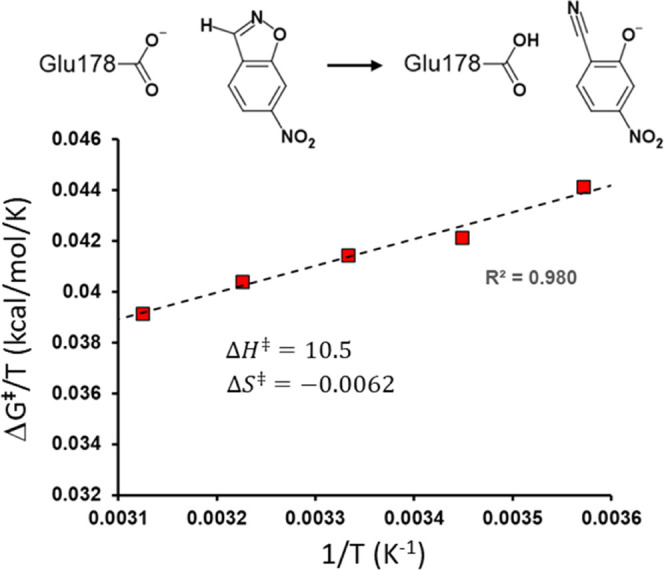
It has been suggested that heat capacity changes in enzyme catalysis may be the underlying reason for temperature optima that are not related to unfolding of the enzyme. If this were to be a common phenomenon, it would have major implications for our interpretation of enzyme kinetics. In most cases, the support for the possible existence of a nonzero (negative) activation heat capacity, however, only relies on fitting such a kinetic model to experimental data. It is therefore of fundamental interest to try to use computer simulations to address this issue. One way is simply to calculate the temperature dependence of the activation free energy and determine whether the relationship is linear or not. An alternative approach is to calculate the absolute heat capacities of the reactant and transition states from plain molecular dynamics simulations using either the temperature derivative or fluctuation formula for the enthalpy. Here, we examine these different approaches for a designer enzyme with a temperature optimum that is not caused by unfolding. Benchmark calculations for the heat capacity of liquid water are first carried out using different thermostats. It is shown that the derivative formula for the heat capacity is generally the most robust and insensitive to the thermostat used and its parameters. The enzyme calculations using this method give results in agreement with direct calculations of activation free energies and show no sign of a negative activation heat capacity. We also provide a simple scheme for the calculation of binding heat capacity changes, which is of clear interest in ligand design, and demonstrate it for substrate binding to the designer enzyme. Neither in that case do the simulations predict any negative heat capacity change.
有人认为,酶催化中的热容变化可能是与酶解折叠无关的温度最适的潜在原因。如果这是一种普遍现象,那么它将对我们对酶动力学的解释产生重大影响。在大多数情况下,对于可能存在非零(负)活化热容的支持,仅依赖于将这种动力学模型拟合到实验数据。因此,尝试使用计算机模拟来解决这个问题具有根本意义。一种方法是简单地计算活化自由能随温度的变化,并确定关系是否线性。另一种方法是使用温度导数或波动公式从纯分子动力学模拟中计算反应物和过渡态的绝对热容。在这里,我们研究了具有非解折叠引起的最适温度的设计酶的这些不同方法。首先使用不同的恒温器对液态水的热容进行基准计算。结果表明,热容的导数公式通常最稳健,对使用的恒温器及其参数不敏感。使用该方法进行的酶计算结果与直接计算的活化自由能一致,并且没有出现负活化热容的迹象。我们还提供了一种用于计算结合热容量变化的简单方案,该方案在配体设计中具有明显的意义,并针对设计酶的底物结合进行了演示。在这两种情况下,模拟都没有预测到任何负的热容变化。