Nancy E. and Peter C. Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY, USA.
Department of Internal Medicine, Makerere University College of Health Sciences, Kampala, Uganda.
Sci Rep. 2022 Oct 10;12(1):16972. doi: 10.1038/s41598-022-21244-x.
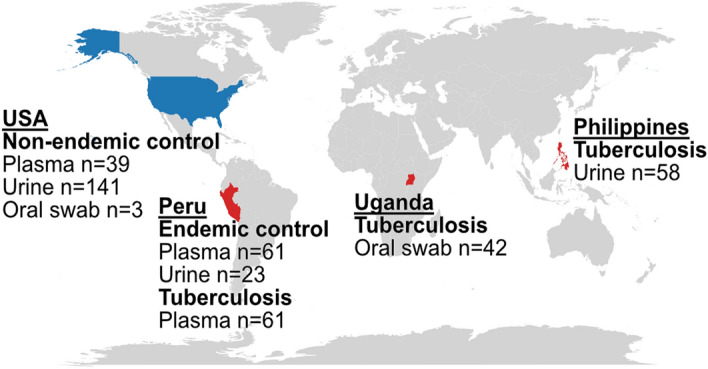
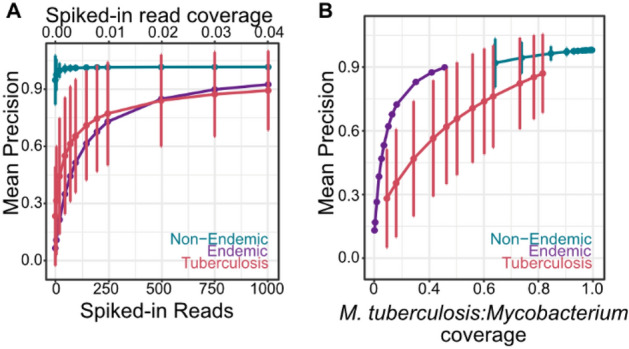
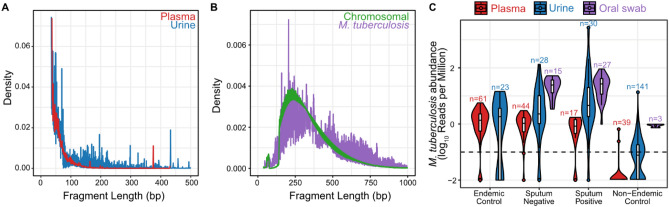
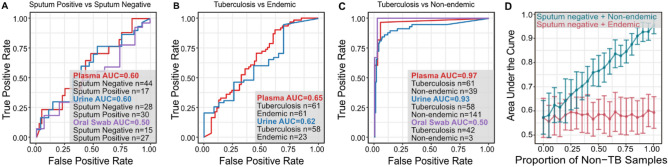
Tuberculosis (TB) remains a significant cause of mortality worldwide. Metagenomic next-generation sequencing has the potential to reveal biomarkers of active disease, identify coinfection, and improve detection for sputum-scarce or culture-negative cases. We conducted a large-scale comparative study of 428 plasma, urine, and oral swab samples from 334 individuals from TB endemic and non-endemic regions to evaluate the utility of a shotgun metagenomic DNA sequencing assay for tuberculosis diagnosis. We found that the composition of the control population had a strong impact on the measured performance of the diagnostic test: the use of a control population composed of individuals from a TB non-endemic region led to a test with nearly 100% specificity and sensitivity, whereas a control group composed of individuals from TB endemic regions exhibited a high background of nontuberculous mycobacterial DNA, limiting the diagnostic performance of the test. Using mathematical modeling and quantitative comparisons to matched qPCR data, we found that the burden of Mycobacterium tuberculosis DNA constitutes a very small fraction (0.04 or less) of the total abundance of DNA originating from mycobacteria in samples from TB endemic regions. Our findings suggest that the utility of a minimally invasive metagenomic sequencing assay for pulmonary tuberculosis diagnostics is limited by the low burden of M. tuberculosis and an overwhelming biological background of nontuberculous mycobacterial DNA.
结核病(TB)仍然是全球范围内导致死亡的重要原因。宏基因组下一代测序有可能揭示活动性疾病的生物标志物,鉴定合并感染,并提高对痰量少或培养阴性病例的检出率。我们对来自结核病流行和非流行地区的 334 名个体的 428 份血浆、尿液和口腔拭子样本进行了大规模的比较研究,以评估全基因组 shotgun 宏基因组 DNA 测序检测在结核病诊断中的应用。我们发现,对照人群的组成对诊断检测的测量性能有很大影响:使用由来自结核病非流行地区的个体组成的对照人群,可使检测具有近 100%的特异性和敏感性,而由来自结核病流行地区的个体组成的对照组则显示出非结核分枝杆菌 DNA 的高背景,限制了检测的诊断性能。我们通过数学建模和与匹配 qPCR 数据的定量比较发现,来自结核病流行地区样本中,结核分枝杆菌 DNA 的负担仅占来自分枝杆菌总 DNA 丰度的很小一部分(0.04 或更少)。我们的研究结果表明,微创宏基因组测序检测在肺结核诊断中的应用受到结核分枝杆菌负担低和非结核分枝杆菌 DNA 的压倒性生物学背景的限制。