Thangavel Neelaveni, Albratty Mohammed
Department of Pharmaceutical Chemistry and Pharmacognosy, College of Pharmacy, Jazan University, Jazan, Saudi Arabia.
Arab J Chem. 2022 Dec;15(12):104334. doi: 10.1016/j.arabjc.2022.104334. Epub 2022 Oct 12.
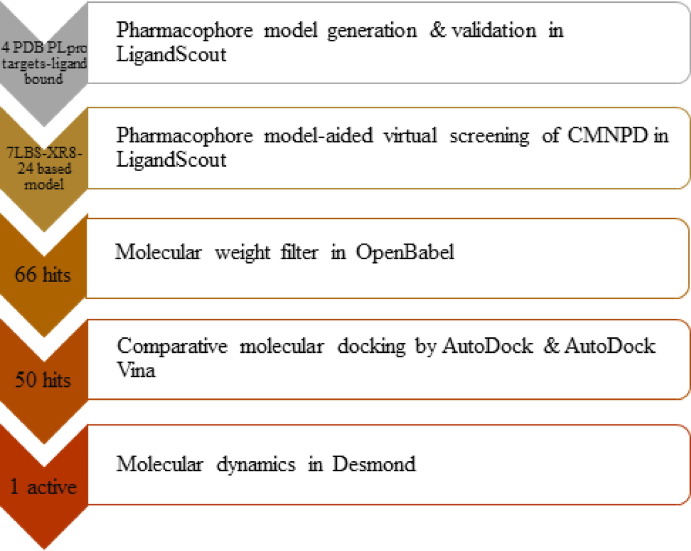
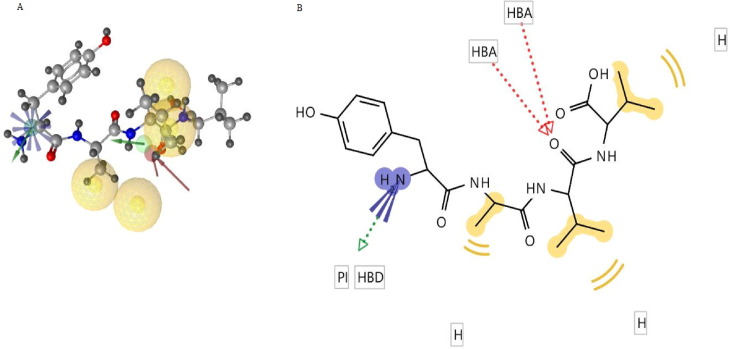
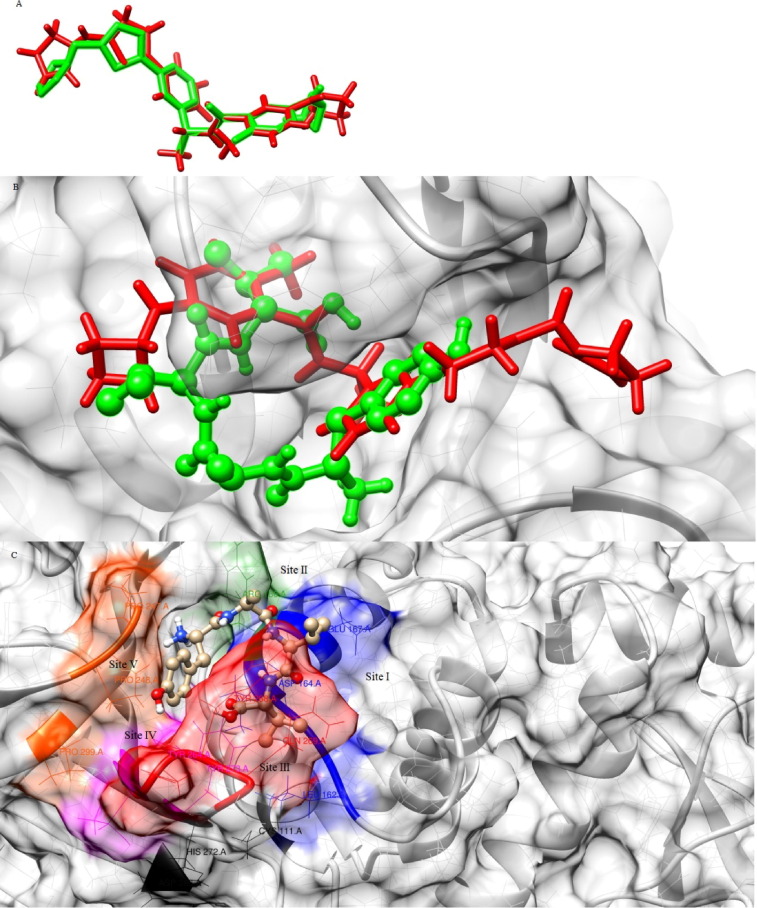
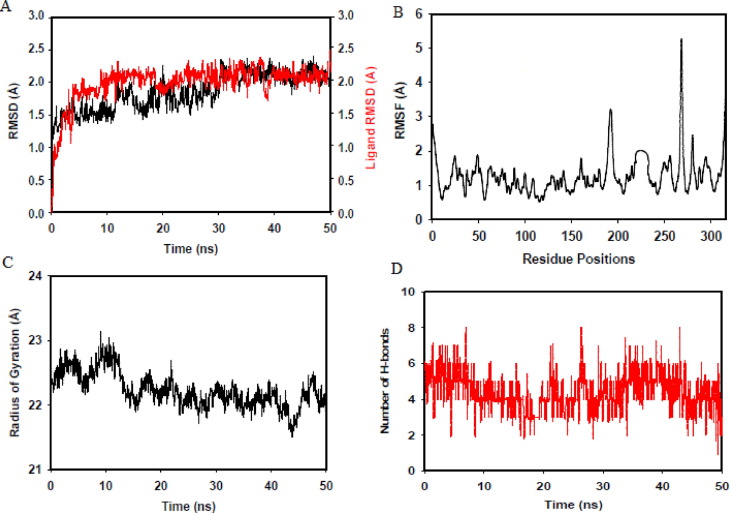
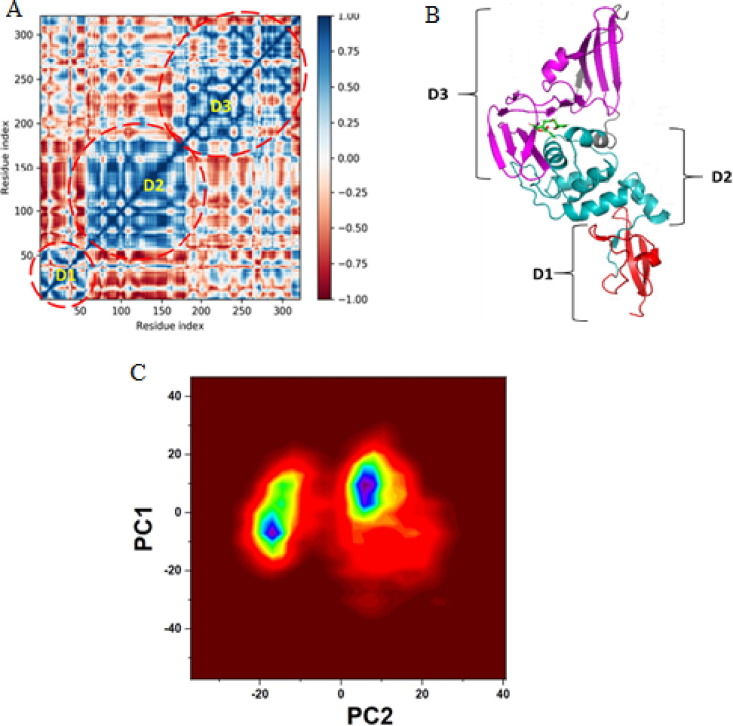
Targeting SARS-CoV-2 papain-like protease using inhibitors is a suitable approach for inhibition of virus replication and dysregulation of host anti-viral immunity. Engaging all five binding sites far from the catalytic site of PLpro is essential for developing a potent inhibitor. We developed and validated a structure-based pharmacophore model with 9 features of a potent PLpro inhibitor. The pharmacophore model-aided virtual screening of the comprehensive marine natural product database predicted 66 initial hits. This hit library was downsized by filtration through a molecular weight filter of ≤ 500 g/mol. The 50 resultant hits were screened by comparative molecular docking using AutoDock and AutoDock Vina. Comparative molecular docking enables benchmarking docking and relieves the disparities in the search and scoring functions of docking engines. Both docking engines retrieved 3 same compounds at different positions in the top 1 % rank, hence consensus scoring was applied, through which CMNPD28766, aspergillipeptide F emerged as the best PLpro inhibitor. Aspergillipeptide F topped the 50-hit library with a pharmacophore-fit score of 75.916. Favorable binding interactions were predicted between aspergillipeptide F and PLpro similar to the native ligand XR8-24. Aspergillipeptide F was able to engage all the 5 binding sites including the newly discovered BL2 groove, site V. Molecular dynamics for quantification of Cα-atom movements of PLpro after ligand binding indicated that it exhibits highly correlated domain movements contributing to the low free energy of binding and a stable conformation. Thus, aspergillipeptide F is a promising candidate for pharmaceutical and clinical development as a potent SARS-CoV-2 PLpro inhibitor.
使用抑制剂靶向严重急性呼吸综合征冠状病毒2(SARS-CoV-2)木瓜样蛋白酶是抑制病毒复制和宿主抗病毒免疫失调的合适方法。占据远离木瓜样蛋白酶(PLpro)催化位点的所有五个结合位点对于开发有效的抑制剂至关重要。我们开发并验证了一个具有有效PLpro抑制剂9个特征的基于结构的药效团模型。通过该药效团模型辅助对综合海洋天然产物数据库进行虚拟筛选,预测出66个初始命中物。通过分子量过滤器(≤500 g/mol)过滤,减少命中物库的数量。使用AutoDock和AutoDock Vina通过比较分子对接对得到的50个命中物进行筛选。比较分子对接能够进行对接基准测试,并消除对接引擎在搜索和评分功能上的差异。两个对接引擎在前1%排名中在不同位置检索到3种相同的化合物,因此应用了一致性评分,据此曲霉肽F(CMNPD28766)成为最佳的PLpro抑制剂。曲霉肽F在50个命中物库中得分最高,药效团拟合分数为75.916。预测曲霉肽F与PLpro之间存在类似于天然配体XR8-24的有利结合相互作用。曲霉肽F能够占据所有5个结合位点,包括新发现的BL2凹槽(位点V)。用于定量配体结合后PLpro的Cα原子运动的分子动力学表明,它表现出高度相关的结构域运动,这有助于低结合自由能和稳定的构象。因此,曲霉肽F作为一种有效的SARS-CoV-2 PLpro抑制剂,是药物和临床开发的有前途的候选物。