Department of Cellular and Molecular Physiology, Penn State College of Medicine, Hershey, Pennsylvania, USA.
Department of Cellular and Molecular Physiology, Penn State College of Medicine, Hershey, Pennsylvania, USA; Department of Ophthalmology, Penn State College of Medicine, Hershey, Pennsylvania, USA.
J Biol Chem. 2022 Dec;298(12):102638. doi: 10.1016/j.jbc.2022.102638. Epub 2022 Oct 26.
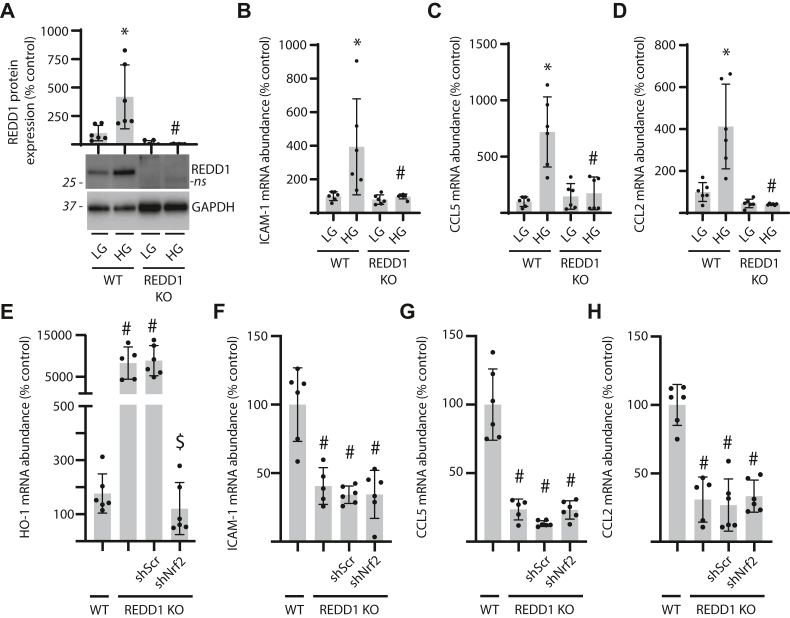
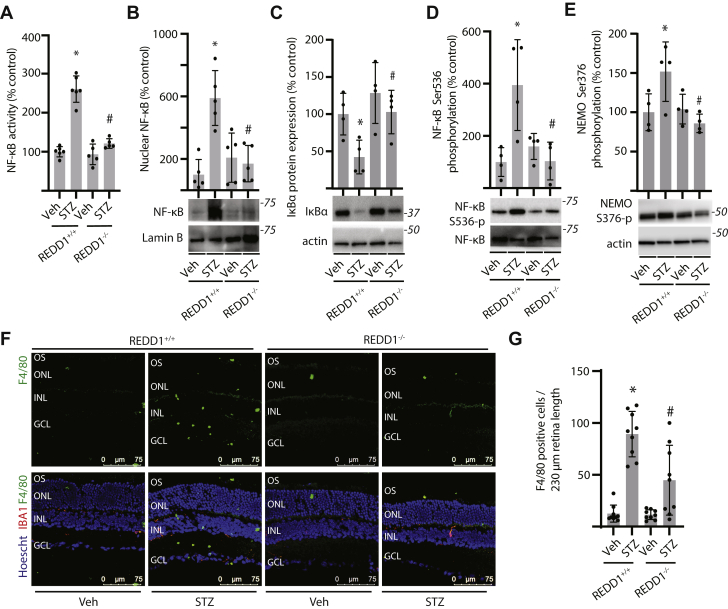
Inflammation contributes to the progression of retinal pathology caused by diabetes. Here, we investigated a role for the stress response protein regulated in development and DNA damage response 1 (REDD1) in the development of retinal inflammation. Increased REDD1 expression was observed in the retina of mice after 16-weeks of streptozotocin (STZ)-induced diabetes, and REDD1 was essential for diabetes-induced pro-inflammatory cytokine expression. In human retinal MIO-M1 Müller cell cultures, REDD1 deletion prevented increased pro-inflammatory cytokine expression in response to hyperglycemic conditions. REDD1 deletion promoted nuclear factor erythroid-2-related factor 2 (Nrf2) hyperactivation; however, Nrf2 was not required for reduced inflammatory cytokine expression in REDD1-deficient cells. Rather, REDD1 enhanced inflammatory cytokine expression by promoting activation of nuclear transcription factor κB (NF-κB). In WT cells exposed to tumor necrosis factor α (TNFα), inflammatory cytokine expression was increased in coordination with activating transcription factor 4 (ATF4)-dependent REDD1 expression and sustained activation of NF-κB. In both Müller cell cultures exposed to TNFα and in the retina of STZ-diabetic mice, REDD1 deletion promoted inhibitor of κB (IκB) expression and reduced NF-κB DNA-binding activity. We found that REDD1 acted upstream of IκB by enhancing both K63-ubiquitination and auto-phosphorylation of IκB kinase complex. In contrast with STZ-diabetic REDD1 mice, IκB kinase complex autophosphorylation and macrophage infiltration were not observed in the retina of STZ-diabetic REDD1 mice. The findings provide new insight into how diabetes promotes retinal inflammation and support a model wherein REDD1 sustains activation of canonical NF-κB signaling.
炎症会导致糖尿病引起的视网膜病变恶化。在这里,我们研究了应激反应蛋白发育调节和 DNA 损伤反应 1(REDD1)在视网膜炎症发展中的作用。在链脲佐菌素(STZ)诱导的糖尿病小鼠的视网膜中观察到 REDD1 表达增加,并且 REDD1 对于糖尿病诱导的促炎细胞因子表达是必需的。在人视网膜 MIO-M1 Müller 细胞培养物中,REDD1 缺失可防止高血糖条件下促炎细胞因子表达增加。REDD1 缺失促进核因子红细胞 2 相关因子 2(Nrf2)过度激活;然而,在 REDD1 缺陷细胞中,Nrf2 对于减少炎症细胞因子表达不是必需的。相反,REDD1 通过促进核转录因子 κB(NF-κB)的激活来增强炎症细胞因子的表达。在暴露于肿瘤坏死因子 α(TNFα)的 WT 细胞中,炎症细胞因子表达增加与激活转录因子 4(ATF4)依赖性 REDD1 表达和 NF-κB 的持续激活协调一致。在暴露于 TNFα 的 Müller 细胞培养物和 STZ 糖尿病小鼠的视网膜中,REDD1 缺失促进了 IκB 表达并降低了 NF-κB DNA 结合活性。我们发现 REDD1 通过增强 IκB 激酶复合物的 K63-泛素化和自身磷酸化来作用于 IκB 的上游。与 STZ 糖尿病 REDD1 小鼠不同,在 STZ 糖尿病 REDD1 小鼠的视网膜中未观察到 IκB 激酶复合物的自身磷酸化和巨噬细胞浸润。这些发现为糖尿病如何促进视网膜炎症提供了新的见解,并支持 REDD1 维持经典 NF-κB 信号转导激活的模型。