Cofas-Vargas Luis Fernando, Mendoza-Espinosa Paola, Avila-Barrientos Luis Pablo, Prada-Gracia Diego, Riveros-Rosas Héctor, García-Hernández Enrique
Universidad Nacional Autónoma de México, Instituto de Química, Ciudad Universitaria, Mexico City, Mexico.
Tecnologico de Monterrey, The Institute for Obesity Research, Monterrey, Mexico.
Front Pharmacol. 2022 Oct 14;13:1012008. doi: 10.3389/fphar.2022.1012008. eCollection 2022.
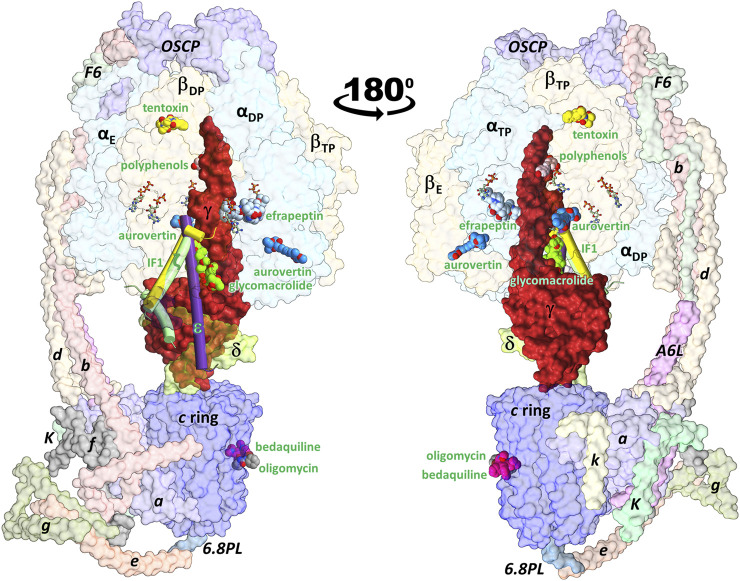
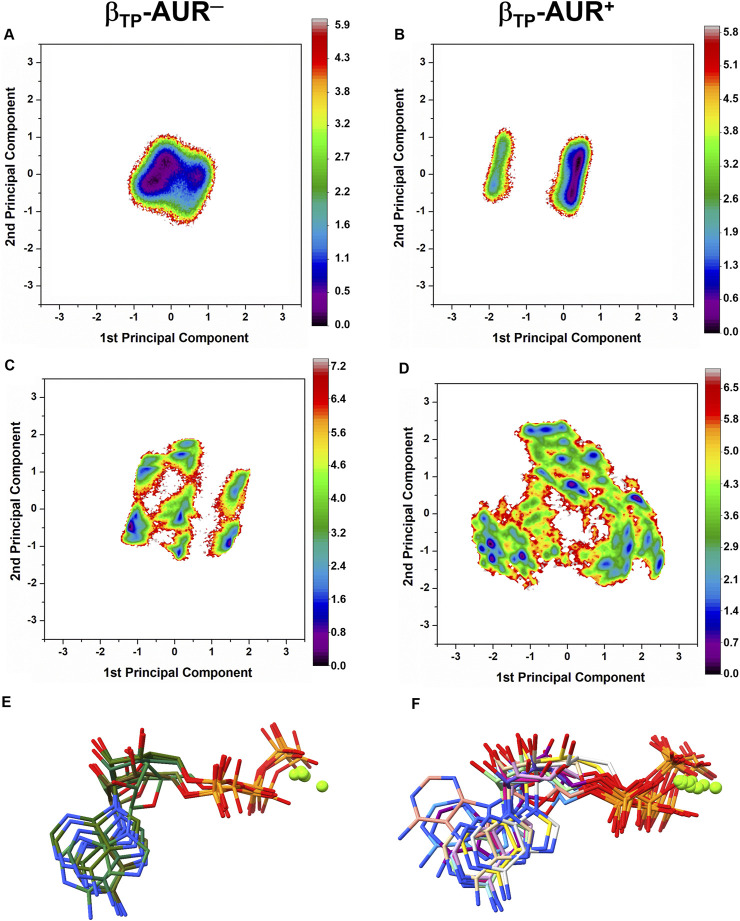
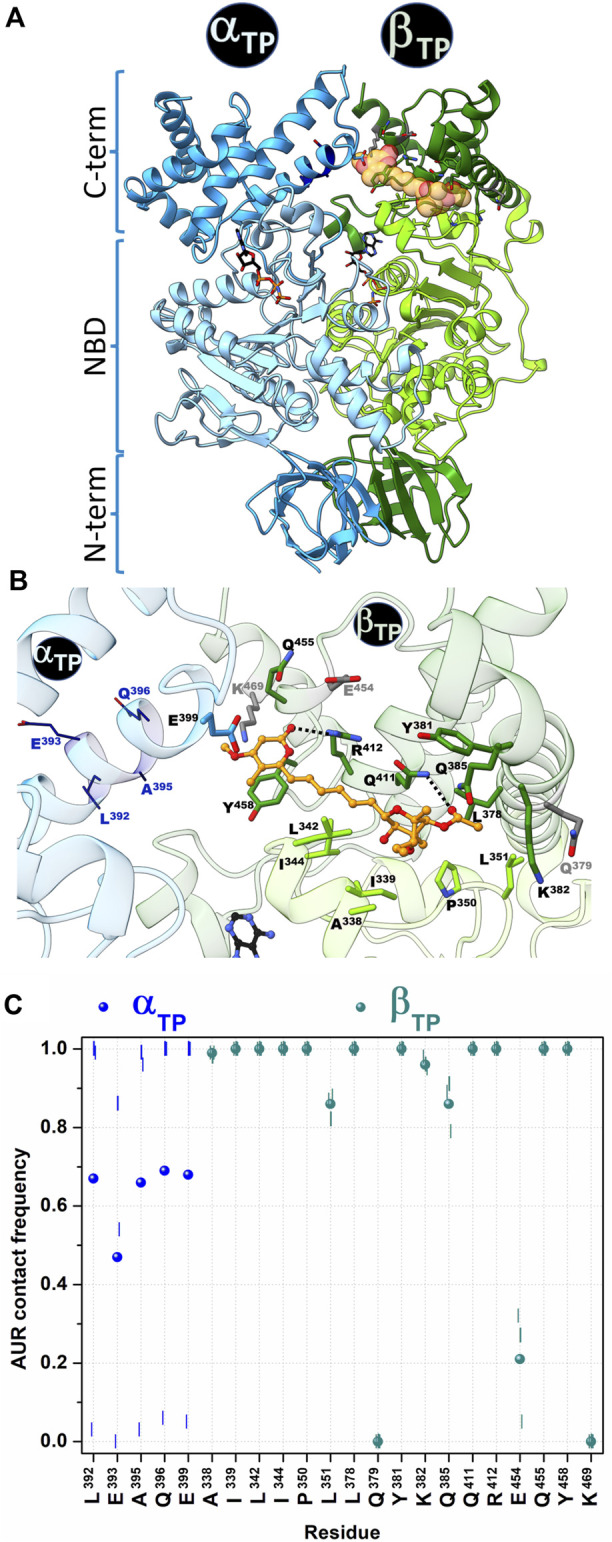
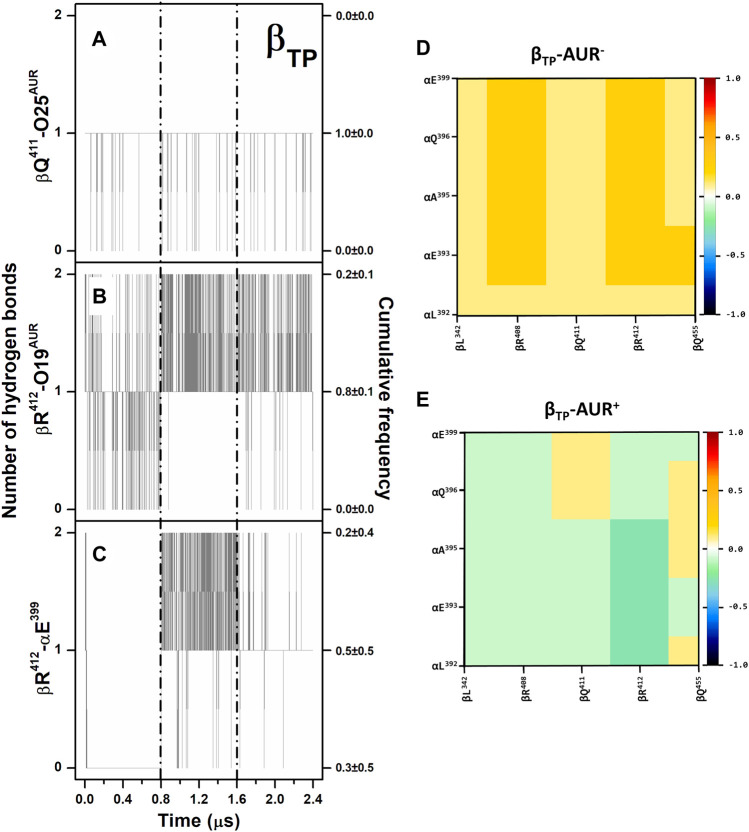
In addition to playing a central role in the mitochondria as the main producer of ATP, FF-ATP synthase performs diverse key regulatory functions in the cell membrane. Its malfunction has been linked to a growing number of human diseases, including hypertension, atherosclerosis, cancer, and some neurodegenerative, autoimmune, and aging diseases. Furthermore, inhibition of this enzyme jeopardizes the survival of several bacterial pathogens of public health concern. Therefore, FF-ATP synthase has emerged as a novel drug target both to treat human diseases and to combat antibiotic resistance. In this work, we carried out a computational characterization of the binding sites of the fungal antibiotic aurovertin in the bovine F subcomplex, which shares a large identity with the human enzyme. Molecular dynamics simulations showed that although the binding sites can be described as preformed, the inhibitor hinders inter-subunit communications and exerts long-range effects on the dynamics of the catalytic site residues. End-point binding free energy calculations revealed hot spot residues for aurovertin recognition. These residues were also relevant to stabilize solvent sites determined from mixed-solvent molecular dynamics, which mimic the interaction between aurovertin and the enzyme, and could be used as pharmacophore constraints in virtual screening campaigns. To explore the possibility of finding species-specific inhibitors targeting the aurovertin binding site, we performed free energy calculations for two bacterial enzymes with experimentally solved 3D structures. Finally, an analysis of bacterial sequences was carried out to determine conservation of the aurovertin binding site. Taken together, our results constitute a first step in paving the way for structure-based development of new allosteric drugs targeting FF-ATP synthase sites of exogenous inhibitors.
除了作为ATP的主要生产者在线粒体中发挥核心作用外,FF-ATP合酶在细胞膜中还执行多种关键的调节功能。其功能失调与越来越多的人类疾病有关,包括高血压、动脉粥样硬化、癌症以及一些神经退行性、自身免疫性和衰老性疾病。此外,抑制这种酶会危及几种引起公共卫生关注的细菌病原体的生存。因此,FF-ATP合酶已成为治疗人类疾病和对抗抗生素耐药性的新型药物靶点。在这项工作中,我们对牛F亚复合体中真菌抗生素奥佛菌素的结合位点进行了计算表征,该亚复合体与人类酶具有很大的同源性。分子动力学模拟表明,虽然结合位点可以描述为预先形成的,但抑制剂会阻碍亚基间的通讯,并对催化位点残基的动力学产生远程影响。终点结合自由能计算揭示了奥佛菌素识别的热点残基。这些残基对于稳定由混合溶剂分子动力学确定的溶剂位点也很重要,混合溶剂分子动力学模拟了奥佛菌素与酶之间的相互作用,并且可以在虚拟筛选活动中用作药效团约束。为了探索寻找靶向奥佛菌素结合位点的物种特异性抑制剂的可能性,我们对两种具有实验解析三维结构的细菌酶进行了自由能计算。最后,对细菌序列进行了分析,以确定奥佛菌素结合位点的保守性。综上所述,我们的结果为基于结构开发针对FF-ATP合酶外源性抑制剂位点的新型变构药物铺平了道路。