Department of Biomedical Engineering, University of Virginia, Charlottesville, VA, 22908, USA.
Department of Microbiology, Immunology & Cancer Biology, University of Virginia, Charlottesville, VA, 22908, USA.
Mol Syst Biol. 2024 Nov;20(11):1230-1256. doi: 10.1038/s44320-024-00064-3. Epub 2024 Sep 27.
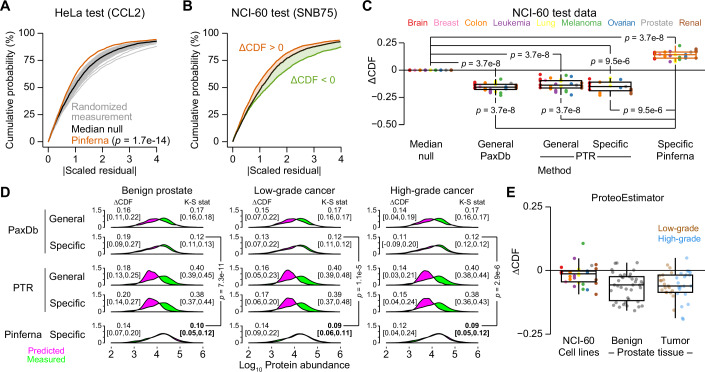
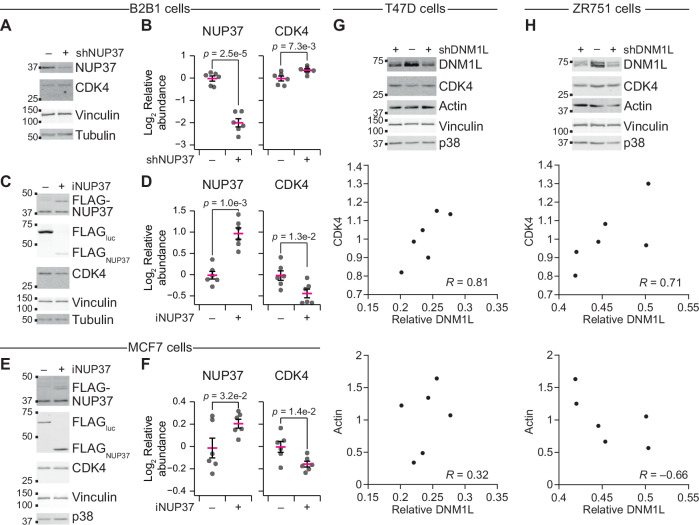
Protein copy numbers constrain systems-level properties of regulatory networks, but proportional proteomic data remain scarce compared to RNA-seq. We related mRNA to protein statistically using best-available data from quantitative proteomics and transcriptomics for 4366 genes in 369 cell lines. The approach starts with a protein's median copy number and hierarchically appends mRNA-protein and mRNA-mRNA dependencies to define an optimal gene-specific model linking mRNAs to protein. For dozens of cell lines and primary samples, these protein inferences from mRNA outmatch stringent null models, a count-based protein-abundance repository, empirical mRNA-to-protein ratios, and a proteogenomic DREAM challenge winner. The optimal mRNA-to-protein relationships capture biological processes along with hundreds of known protein-protein complexes, suggesting mechanistic relationships. We use the method to identify a viral-receptor abundance threshold for coxsackievirus B3 susceptibility from 1489 systems-biology infection models parameterized by protein inference. When applied to 796 RNA-seq profiles of breast cancer, inferred copy-number estimates collectively re-classify 26-29% of luminal tumors. By adopting a gene-centered perspective of mRNA-protein covariation across different biological contexts, we achieve accuracies comparable to the technical reproducibility of contemporary proteomics.
蛋白质拷贝数限制了调控网络的系统级特性,但与 RNA-seq 相比,比例蛋白组学数据仍然很少。我们使用定量蛋白质组学和转录组学中可获得的最佳数据,从 369 个细胞系中的 4366 个基因统计上相关联 mRNA 和蛋白质。该方法从蛋白质的中位数拷贝数开始,并分层附加 mRNA-蛋白质和 mRNA-mRNA 依赖性,以定义一个最佳的基因特异性模型,将 mRNA 与蛋白质联系起来。对于数十个细胞系和原代样本,这些来自 mRNA 的蛋白质推断优于严格的零模型、基于计数的蛋白质丰度存储库、经验 mRNA-蛋白质比和蛋白质基因组学 DREAM 挑战赛的获胜者。最佳的 mRNA-蛋白质关系可以捕捉到生物过程以及数百个已知的蛋白质-蛋白质复合物,这表明存在着机制关系。我们使用该方法从 1489 个基于蛋白质推断的系统生物学感染模型中确定了柯萨奇病毒 B3 易感性的病毒受体丰度阈值。当应用于 796 个乳腺癌的 RNA-seq 图谱时,推断的拷贝数估计值共同重新分类了 26-29%的 luminal 肿瘤。通过在不同的生物背景下采用基因中心的 mRNA-蛋白质共变观点,我们实现了与当代蛋白质组学技术重现性相当的准确性。